Clear Sky Science · pl
Kardiomiocytarne TGFB3 łagodzi włóknienie serca i zachowuje funkcję serca w niewydolności
Dlaczego to ma znaczenie dla osób z osłabionym sercem
Niewydolność serca to powszechna i poważna choroba, w której serce stopniowo traci zdolność do pompowania krwi. Głównym sprawcą jest bliznowacenie wewnątrz serca, zwane włóknieniem, które usztywnia mięsień sercowy i pogarsza objawy. Badanie to ujawnia zaskakujący, ochronny sygnał wytwarzany przez własne komórki mięśniowe serca, który może spowalniać ten proces bliznowacenia i pomagać zachować funkcję serca, wskazując drogę do nowego, bardziej precyzyjnego leczenia niewydolności.
Bliższe spojrzenie na bliznowacenie serca
W niewydolności serca tkanka nie „zużywa się” po prostu. Zamiast tego przebudowuje się: komórki mięśniowe powiększają się lub obumierają, a komórki tkanki łącznej odkładają nadmiar kolagenu, tworząc twarde, bliznopodobne obszary. Od lat potężna rodzina cząsteczek sygnałowych znana jako TGF-beta łączy się z tym procesem bliznowacenia. Blokowanie wszystkich form TGF-beta okazało się jednak zbyt nieprecyzyjne i powodowało poważne skutki uboczne, ponieważ te cząsteczki uczestniczą też w normalnej naprawie i równowadze immunologicznej. Nowe badanie pyta, czy jeden z członków rodziny, zwany TGF-beta3, może w rzeczywistości pomagać sercu bronić się przed wymykającym się włóknieniem, zamiast je napędzać.
Wykrycie ochronnego sygnału w chorych sercach
Naukowcy przeanalizowali tkankę serca od pacjentów z zaawansowaną niewydolnością i od osób bez niewydolności, a także kilka modeli mysich, które naśladują przeciążenie ciśnieniowe serca. Stwierdzili, że poziomy TGF-beta3 były konsekwentnie wyższe w sercach z niewydolnością niż w zdrowych, zarówno w samej tkance, jak i we krwi. Co ważne, wzrost ten nie pochodził głównie od zwykłych komórek tworzących blizny, lecz od kardiomiocytów — komórek mięśniowych, które wykonują skurcze pompujące krew. Pacjenci z wyższymi poziomami TGF-beta3 we krwi mieli też zwykle wyższe stężenia standardowego markera niewydolności serca, co sugeruje, że ta cząsteczka odzwierciedla natężenie obciążenia serca.
Wyłączenie sygnału w komórkach mięśnia sercowego
Aby sprawdzić, czy TGF-beta3 pochodzące z kardiomiocytów jest pomocne czy szkodliwe, zespół skonstruował myszy, w których gen dla TGF-beta3 mógł być selektywnie usunięty tylko w komórkach mięśni serca. W normalnych, spoczynkowych warunkach serca tych myszy wyglądały i funkcjonowały podobnie jak ich współrodzeńcy. Jednak gdy zwierzęta zostały poddane zabiegowi zwężenia głównej tętnicy opuszczającej serce — standardowej metodzie wywoływania przewlekłego przeciążenia ciśnieniowego i ostatecznej niewydolności — różnice stały się uderzające. Myszy pozbawione TGF-beta3 w kardiomiocytach rozwijały gorszą funkcję pompowania, większe serca i znacznie więcej odkładania kolagenu między włóknami mięśni niż zwierzęta kontrolne narażone na ten sam stres.
Jak komórki mięśnia sercowego komunikują się z komórkami tworzącymi blizny
Badając mechanizm, naukowcy porównali aktywność genów w sercach z i bez kardiomiocytarnego TGF-beta3. Serce pozbawione tego sygnału wykazywało gwałtowny wzrost dwóch dobrze znanych czynników napędzających włóknienie, CTGF i SERPINE1, które są produkowane głównie przez fibroblasty — komórki odpowiedzialne za wytwarzanie tkanki łącznej. W eksperymentach na hodowlach komórkowych, gdy fibroblasty były narażone na klasyczny sygnał bliznowacenia TGF-beta1, uruchamiały te geny fibrotczne i przyjmowały aktywowany, produkujący macierz stan. Dodanie TGF-beta3 do tego układu jednak osłabiało tę odpowiedź. Fibroblasty zanurzone w płynie pochodzącym od kardiomiocytów pozbawionych TGF-beta3 stały się bardziej aktywowane niż te wystawione na płyn od normalnych kardiomiocytów, co potwierdza, że komórki mięśniowe zwykle wydzielają czynnik powstrzymujący nadmierną aktywność fibroblastów.
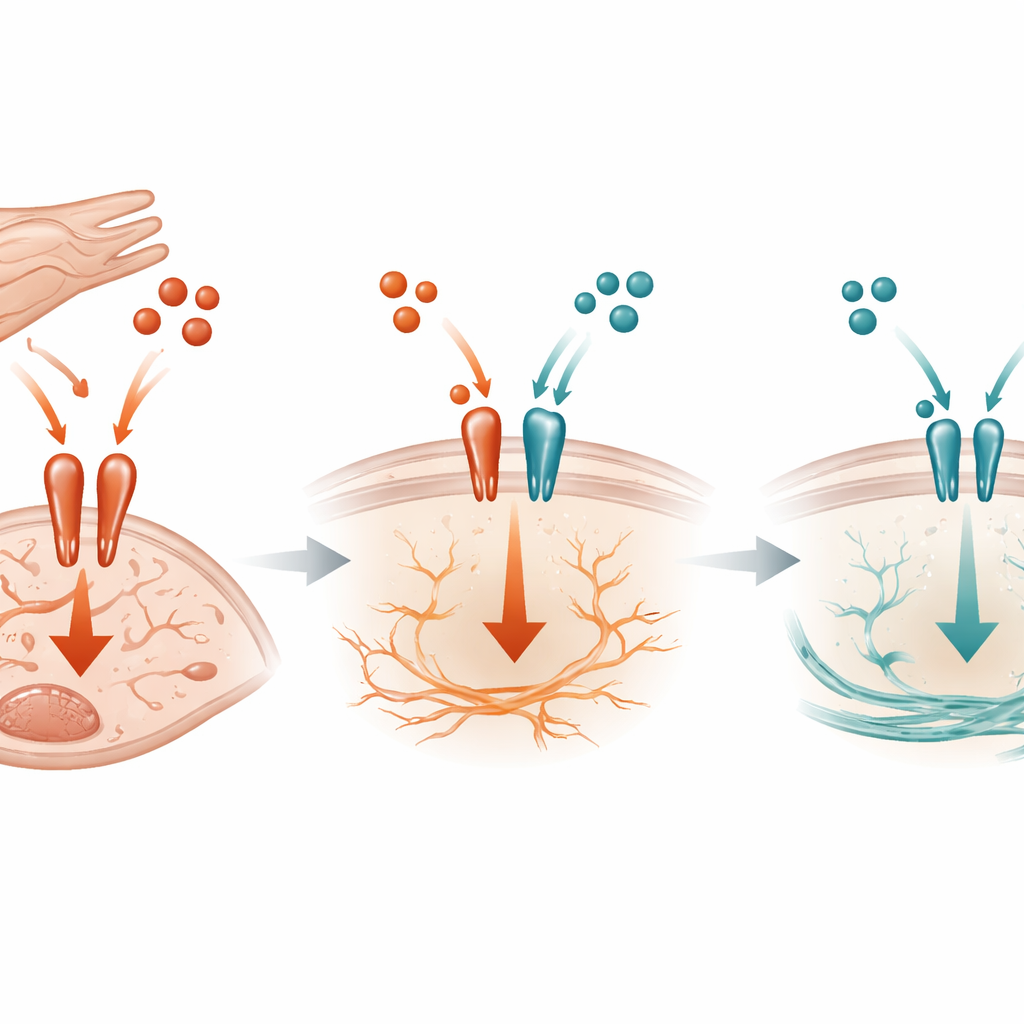
Molekularna przepychanka na powierzchni komórki
Na poziomie molekularnym badanie pokazuje, że TGF-beta3 działa jak konkurencyjny hamulec wobec bardziej agresywnego sygnału TGF-beta1. Obie cząsteczki korzystają z tego samego kompleksu receptorowego na powierzchni fibroblastów, by uruchomić aktywność wewnątrz komórki. Eksperymenty biochemiczne wykazały, że TGF-beta3 może wiązać się z tymi receptorami i ograniczać, ile TGF-beta1 może się do nich przyłączyć, zmniejszając w ten sposób aktywację kluczowego przełącznika wewnątrzkomórkowego zwanego SMAD3. Gdy kardiomiocytarne TGF-beta3 jest nieobecne u myszy pod przeciążeniem ciśnieniowym, aktywacja SMAD3 wzrasta, CTGF i SERPINE1 rosną, a włóknienie przyspiesza. Co istotne, efekt ten nie opierał się na innym znanym inhibitorze w tej ścieżce, co sugeruje, że prosta konkurencja o receptor stanowi kluczową część mechanizmu ochronnego.
Co to może oznaczać dla przyszłych terapii
Dla laika kluczowy przekaz jest taki, że serce w niewydolności nie jest tylko ofiarą bliznowacenia; stara się też chronić samo. Komórki mięśniowe serca uwalniają TGF-beta3 jako lokalną, wbudowaną obronę, która przeciwdziała silniejszym sygnałom pro-bliznowatym i powstrzymuje tkankę łączną przed nadmierną aktywacją. Gdy ten ochronny sygnał jest nieobecny, włóknienie się nasila, a funkcja serca pogarsza się szybciej. Zamiast blokować cały system TGF-beta, przyszłe terapie mogłyby dążyć do wzmocnienia lub naśladowania TGF-beta3 w sercu, wzmacniając własną zdolność narządu do ograniczania szkodliwego bliznowacenia przy jednoczesnym zachowaniu potrzebnej naprawy w innych tkankach.
Cytowanie: Xuan, J., Zhou, J., Huang, Y. et al. Cardiomyocyte-derived TGFB3 attenuates cardiac fibrosis and preserves cardiac function in heart failure. Sci Rep 16, 11534 (2026). https://doi.org/10.1038/s41598-026-42367-5
Słowa kluczowe: niewydolność serca, włóknienie serca, TGF-beta3, kardiomiocyty, sygnalizacja fibroblastów