Clear Sky Science · it
La TGFB3 derivata dai cardiomiociti attenua la fibrosi cardiaca e preserva la funzione cardiaca nell’insufficienza cardiaca
Perché questo è importante per le persone con cuori deboli
L’insufficienza cardiaca è una condizione comune e grave in cui il cuore perde progressivamente la capacità di pompare sangue. Un colpevole importante è la formazione di cicatrici all’interno del cuore, chiamata fibrosi, che irrigidisce il muscolo cardiaco e peggiora i sintomi. Questo studio scopre un segnale protettivo sorprendente prodotto dalle stesse cellule muscolari del cuore che può rallentare questo processo di cicatrizzazione e contribuire a preservare la funzione cardiaca, indicando una nuova strada più mirata per trattare l’insufficienza cardiaca.
Uno sguardo più attento alla cicatrizzazione cardiaca
Nell’insufficienza cardiaca, il cuore non si limita a “consumarsi”. Il tessuto si rimodella: i miociti aumentano di volume o muoiono, e le cellule del tessuto connettivo depositano collagene in eccesso, formando aree rigide simili a cicatrici. Per anni una potente famiglia di molecole segnale nota come TGF-beta è stata collegata a questa cicatrizzazione. Bloccare tutte le forme di TGF-beta, tuttavia, si è rivelato uno strumento troppo grossolano, causando effetti collaterali seri perché queste molecole partecipano anche alla riparazione normale e all’equilibrio immunitario. Il nuovo lavoro si chiede se un membro particolare di questa famiglia, chiamato TGF-beta3, possa in realtà aiutare il cuore a difendersi dalla fibrosi incontrollata invece di favorirla.
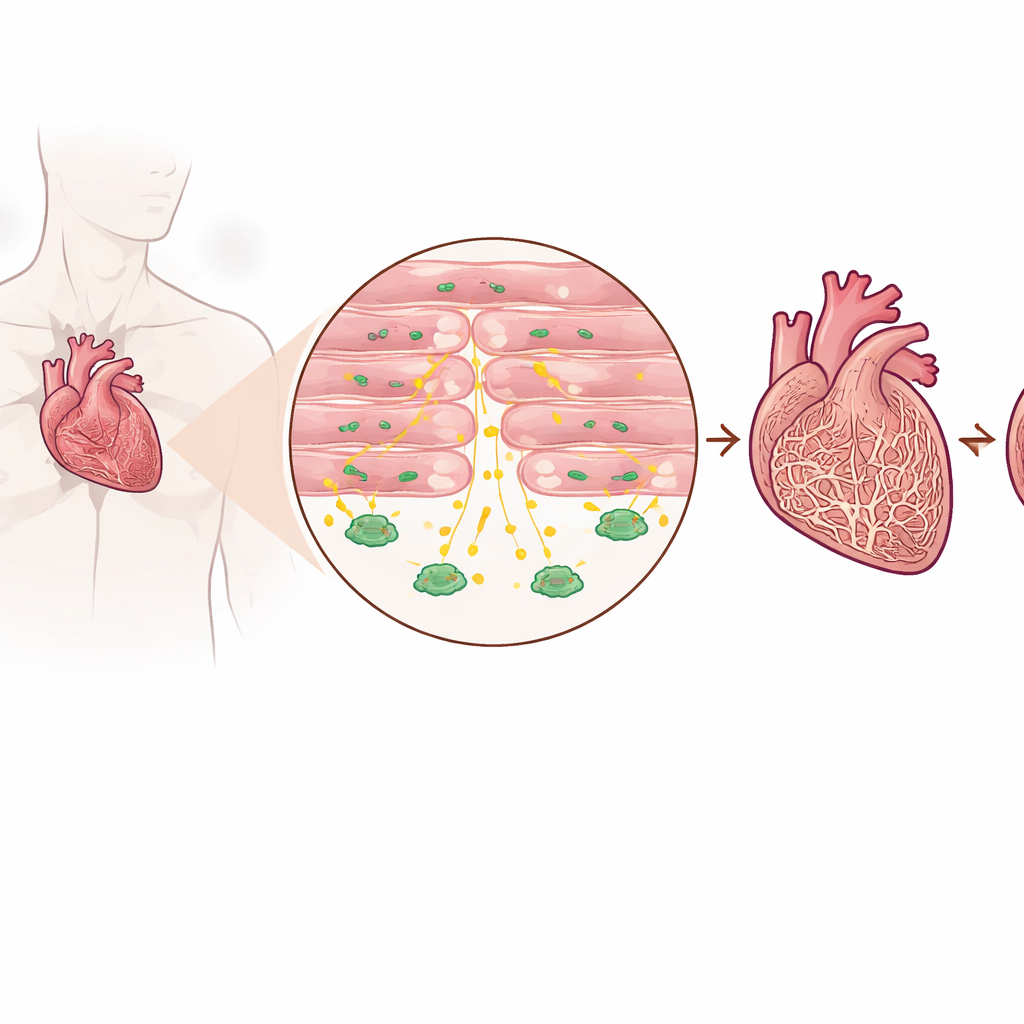
Rilevare un segnale protettivo nei cuori malati
I ricercatori hanno esaminato tessuto cardiaco di pazienti con insufficienza cardiaca avanzata e di persone senza insufficienza cardiaca, oltre a diversi modelli murini che riproducono il sovraccarico di pressione sul cuore. Hanno scoperto che i livelli di TGF-beta3 erano costantemente più alti nei cuori in insufficienza rispetto a quelli sani, sia nel tessuto sia nel flusso sanguigno. È importante che questo aumento non provenisse principalmente dalle consuete cellule che formano cicatrici, ma dai cardiomiociti — le cellule muscolari che si contraggono per pompare il sangue. I pazienti con livelli ematici più elevati di TGF-beta3 tendevano inoltre ad avere livelli più alti di un marcatore standard dell’insufficienza cardiaca, suggerendo che questa molecola rifletta l’intensità dello stress a cui è sottoposto il cuore.
Spegnere il segnale nelle cellule muscolari cardiache
Per verificare se il TGF-beta3 prodotto dai cardiomiociti fosse utile o dannoso, il gruppo ha ingegnerizzato topi in cui il gene per TGF-beta3 poteva essere eliminato selettivamente solo nelle cellule muscolari cardiache. In condizioni normali e di riposo, questi topi avevano cuori dall’aspetto e dalla funzione simili a quelli dei loro avoli. Ma quando gli animali sono stati sottoposti a una procedura che restringe l’arteria principale che esce dal cuore — un metodo standard per indurre sovraccarico cronico di pressione e insufficienza cardiaca — le differenze sono diventate evidenti. I topi privi di TGF-beta3 nei cardiomiociti hanno sviluppato una funzione di pompaggio peggiore, cuori più grandi e un accumulo molto maggiore di collagene tra le fibre muscolari rispetto agli animali di controllo esposti allo stesso stress.
Come le cellule muscolari cardiache comunicano con le cellule che formano la cicatrice
Approfondendo il meccanismo, i ricercatori hanno confrontato l’attività genica nei cuori con e senza TGF-beta3 nei cardiomiociti. I cuori privi di questo segnale mostravano un netto aumento di due noti promotori della fibrosi, CTGF e SERPINE1, prodotti principalmente dai fibroblasti, le cellule responsabili della produzione del tessuto connettivo. In esperimenti in coltura cellulare, quando i fibroblasti venivano esposti al classico segnale di cicatrizzazione TGF-beta1, attivavano questi geni fibrotici e assumevano uno stato attivato produttrice di matrice. Aggiungere TGF-beta3 alla miscela, tuttavia, attenuava questa risposta. I fibroblasti immersi nel fluido proveniente da cardiomiociti privi di TGF-beta3 diventavano più attivati rispetto a quelli esposti al fluido da cardiomiociti normali, confermando che le cellule muscolari normalmente secernono un fattore che frena l’iperattività dei fibroblasti.
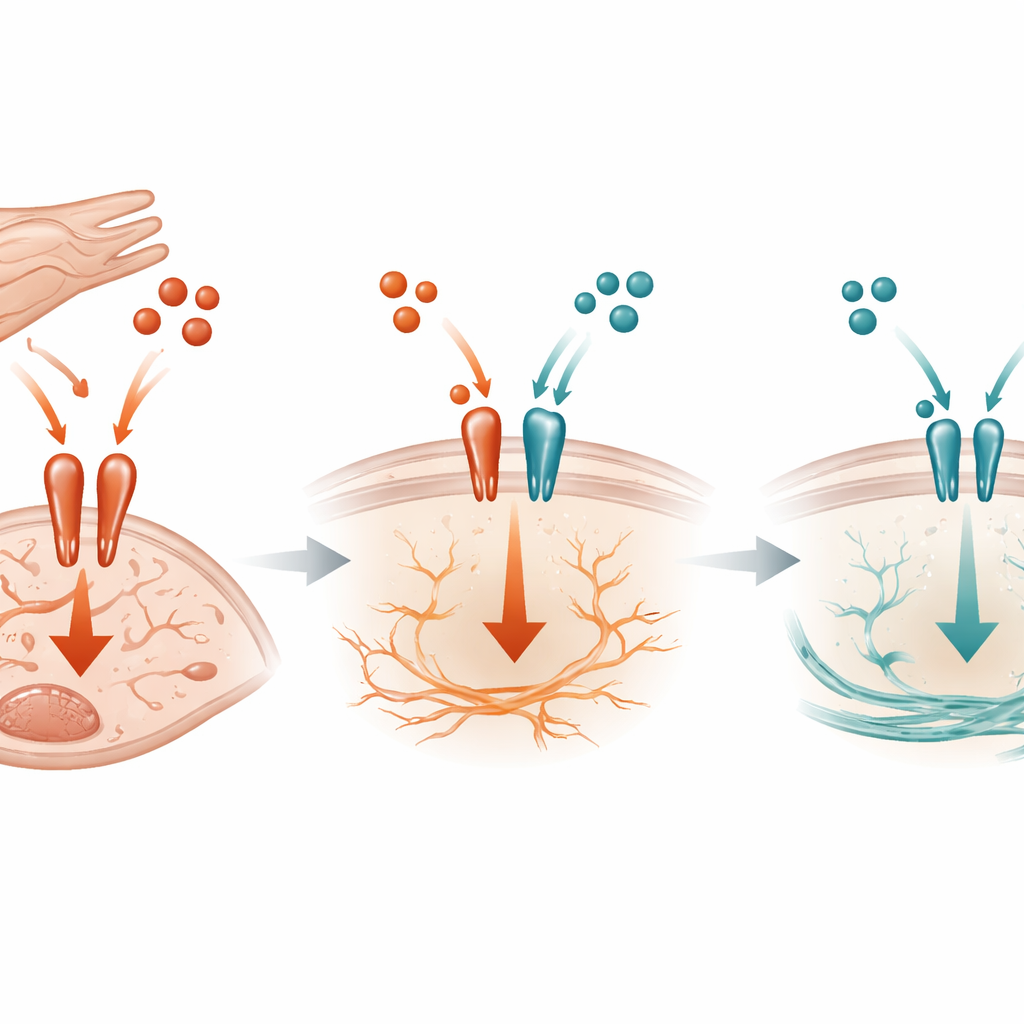
Una contesa molecolare sulla superficie cellulare
A livello molecolare, lo studio mostra che il TGF-beta3 agisce come un freno competitivo sul più aggressivo segnale TGF-beta1. Entrambe le molecole usano lo stesso complesso recettoriale sulla superficie dei fibroblasti per innescare l’attività all’interno della cellula. Esperimenti biochimici hanno rivelato che il TGF-beta3 può legarsi a questi recettori e limitare quanto TGF-beta1 può attaccarsi, riducendo così l’attivazione di un interruttore chiave all’interno della cellula chiamato SMAD3. Quando il TGF-beta3 derivato dai cardiomiociti è assente nei topi sottoposti a sovraccarico di pressione, l’attivazione di SMAD3 aumenta, CTGF e SERPINE1 crescono e la fibrosi accelera. È notevole che questo effetto non dipendesse da un altro noto inibitore all’interno della via, suggerendo che la semplice competizione per il recettore sia una parte centrale del meccanismo protettivo.
Cosa potrebbe significare per i trattamenti futuri
Per un non specialista, il messaggio chiave è che il cuore in insufficienza non è solo una vittima della cicatrizzazione; cerca anche di proteggersi. Le cellule muscolari cardiache rilasciano TGF-beta3 come una difesa locale e innata che si contrappone a segnali più forti pro-cicatrizzazione e impedisce al tessuto connettivo di entrare in iperattivazione. Quando questo segnale protettivo manca, la fibrosi peggiora e la funzione cardiaca declina più rapidamente. Piuttosto che bloccare l’intero sistema TGF-beta, le terapie future potrebbero mirare ad aumentare o imitare specificamente il TGF-beta3 nel cuore, potenziando la capacità dell’organo di limitare la cicatrizzazione dannosa preservando la riparazione necessaria in altri tessuti.
Citazione: Xuan, J., Zhou, J., Huang, Y. et al. Cardiomyocyte-derived TGFB3 attenuates cardiac fibrosis and preserves cardiac function in heart failure. Sci Rep 16, 11534 (2026). https://doi.org/10.1038/s41598-026-42367-5
Parole chiave: insufficienza cardiaca, fibrosi cardiaca, TGF-beta3, cardiomiociti, segnalazione dei fibroblasti