Clear Sky Science · en
Cardiomyocyte-derived TGFB3 attenuates cardiac fibrosis and preserves cardiac function in heart failure
Why this matters for people with weak hearts
Heart failure is a common and serious condition in which the heart gradually loses its ability to pump blood. A major culprit is scarring inside the heart, called fibrosis, which stiffens the heart muscle and worsens symptoms. This study uncovers a surprising protective signal made by the heart’s own muscle cells that can slow this scarring process and help preserve heart function, pointing toward a new, more precise way to treat heart failure.
A closer look at heart scarring
In heart failure, the heart does not simply “wear out.” Instead, its tissue remodels: muscle cells enlarge or die, and connective tissue cells lay down excess collagen, forming tough scar-like areas. For years, a powerful family of signaling molecules known as TGF-beta has been linked to this scarring. Blocking all forms of TGF-beta, however, proved too blunt an instrument, causing serious side effects because these molecules also help with normal repair and immune balance. The new work asks whether one particular family member, called TGF-beta3, might actually help the heart defend itself against runaway fibrosis rather than drive it.
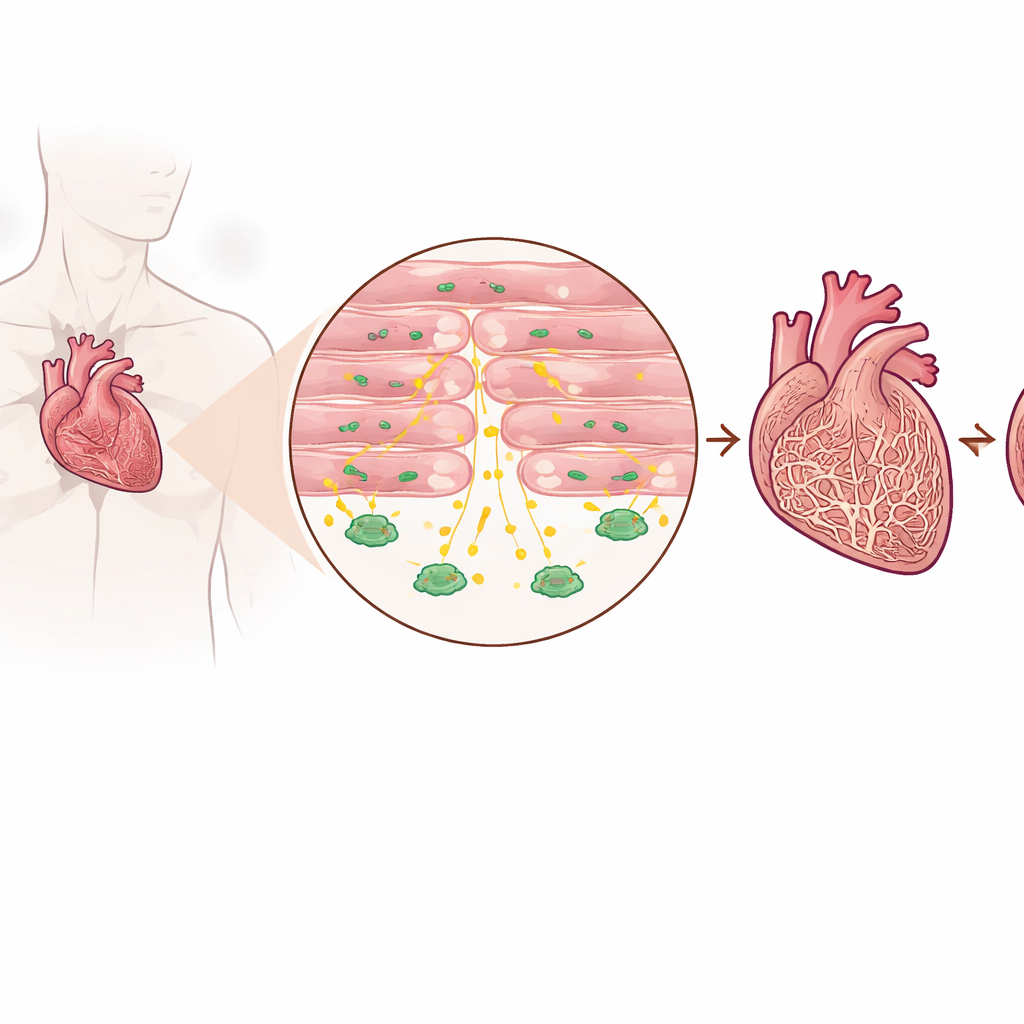
Detecting a protective signal in sick hearts
The researchers examined heart tissue from patients with advanced heart failure and from people without heart failure, as well as several mouse models that mimic pressure overload on the heart. They found that TGF-beta3 levels were consistently higher in failing hearts than in healthy ones, both in the tissue itself and in the bloodstream. Importantly, this increase did not come mainly from the usual scar-forming cells, but from cardiomyocytes—the muscle cells that actually contract to pump blood. Patients with higher blood levels of TGF-beta3 also tended to have higher levels of a standard heart failure marker, suggesting that this molecule reflects the intensity of stress the heart is under.
Switching off the signal in heart muscle cells
To test whether this cardiomyocyte-made TGF-beta3 was helpful or harmful, the team engineered mice in which the gene for TGF-beta3 could be selectively deleted only in heart muscle cells. Under normal, resting conditions, these mice had hearts that looked and functioned much like those of their littermates. But when the animals were subjected to a procedure that constricts the main artery leaving the heart—a standard way to induce chronic pressure overload and eventual heart failure—the differences became striking. Mice lacking TGF-beta3 in their cardiomyocytes developed worse pumping function, larger hearts, and far more collagen buildup between muscle fibers than control animals exposed to the same stress.
How heart muscle cells talk to scar-forming cells
Digging into the mechanism, the researchers compared gene activity in hearts with and without cardiomyocyte TGF-beta3. Hearts missing this signal showed a sharp increase in two well-known drivers of fibrosis, CTGF and SERPINE1, which are produced mainly by fibroblasts, the cells responsible for making connective tissue. In cell culture experiments, when fibroblasts were exposed to the classic scarring signal TGF-beta1, they turned on these fibrotic genes and adopted an activated, matrix-producing state. Adding TGF-beta3 to the mix, however, dampened this response. Fibroblasts bathed in fluid from TGF-beta3-deficient cardiomyocytes became more activated than those exposed to fluid from normal cardiomyocytes, confirming that the muscle cells normally secrete a factor that restrains fibroblast overactivity.
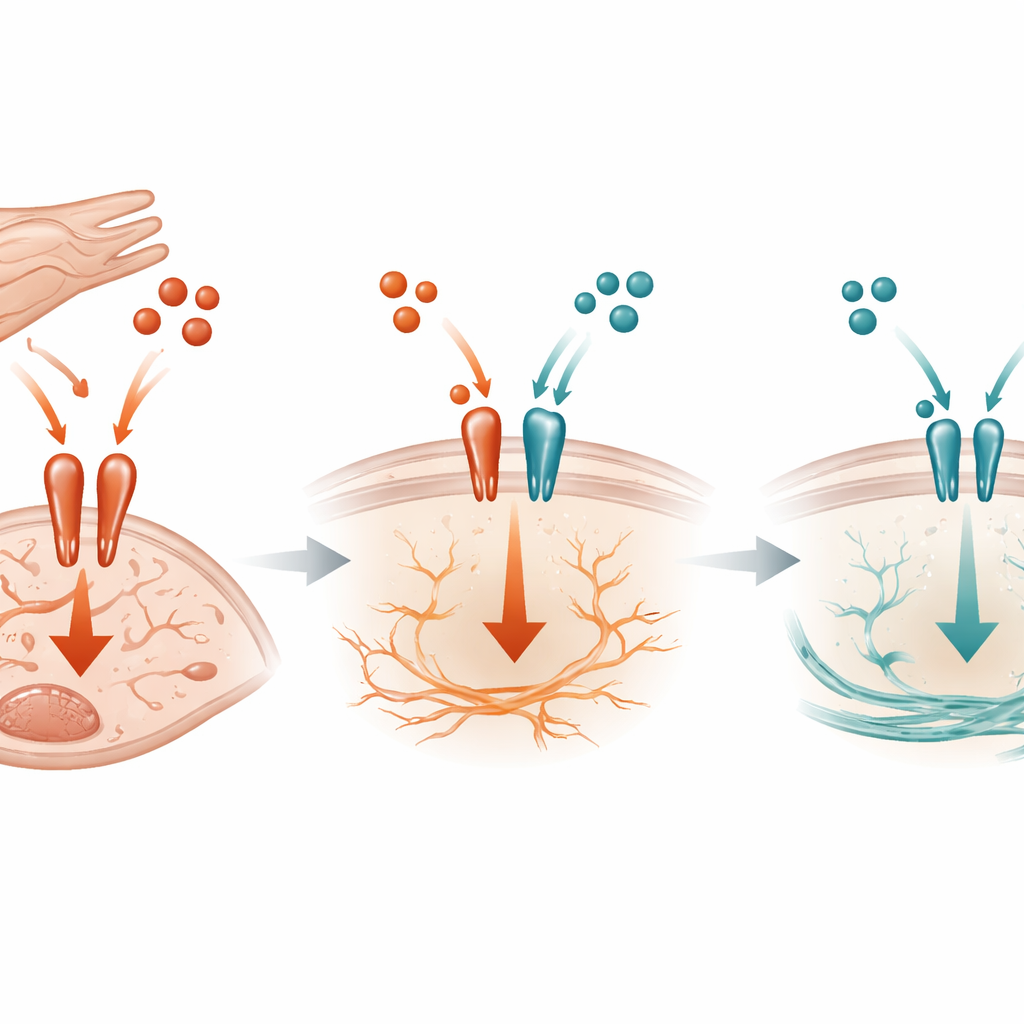
A molecular tug-of-war at the cell surface
At the molecular level, the study shows that TGF-beta3 acts like a competitive brake on the more aggressive TGF-beta1 signal. Both molecules use the same receptor complex on fibroblast surfaces to trigger activity inside the cell. Biochemical experiments revealed that TGF-beta3 can bind to these receptors and limit how much TGF-beta1 can latch on, thereby reducing activation of a key switch inside the cell called SMAD3. When cardiomyocyte-derived TGF-beta3 is absent in mice under pressure overload, SMAD3 activation rises, CTGF and SERPINE1 increase, and fibrosis accelerates. Notably, this effect did not rely on another known inhibitor within the pathway, suggesting that simple competition for the receptor is a central part of the protective mechanism.
What this could mean for future treatments
To a non-specialist, the key message is that the failing heart is not only a victim of scarring; it also tries to protect itself. Heart muscle cells release TGF-beta3 as a local, built-in defense that tugs against stronger pro-scarring signals and keeps connective tissue from going into overdrive. When this protective signal is missing, fibrosis worsens and heart function declines more rapidly. Rather than blocking the entire TGF-beta system, future therapies might aim to boost or mimic TGF-beta3 specifically in the heart, enhancing the organ’s own ability to limit harmful scarring while preserving needed healing in other tissues.
Citation: Xuan, J., Zhou, J., Huang, Y. et al. Cardiomyocyte-derived TGFB3 attenuates cardiac fibrosis and preserves cardiac function in heart failure. Sci Rep 16, 11534 (2026). https://doi.org/10.1038/s41598-026-42367-5
Keywords: heart failure, cardiac fibrosis, TGF-beta3, cardiomyocytes, fibroblast signaling