Clear Sky Science · ru
ТГФБ3, выделяемый кардиомиоцитами, ослабляет сердечный фиброз и сохраняет функцию сердца при сердечной недостаточности
Почему это важно для людей с ослабленным сердцем
Сердечная недостаточность — распространённое и тяжёлое состояние, при котором сердце постепенно теряет способность перекачивать кровь. Одной из главных причин является рубцевание внутри сердца, называемое фиброзом: оно делает мышцу жёсткой и усугубляет симптомы. В этом исследовании обнаружен неожиданный защитный сигнал, вырабатываемый самими кардиомиоцитами, который может замедлять процесс образования рубцов и помогать сохранять функцию сердца, указывая на новый, более точный подход к лечению сердечной недостаточности.
Ближе к механике рубцевания сердца
При сердечной недостаточности сердце не просто «изнашивается». Его ткань перестраивается: мышечные клетки увеличиваются в размерах или погибают, а клетки соединительной ткани откладывают избыток коллагена, формируя жёсткие подобия рубцов. На протяжении многих лет сильная семейству сигнальных молекул, известных как TGF‑beta, приписывали роль в этом рубцевании. Однако блокирование всех форм TGF‑beta оказалось слишком грубым инструментом и вызывало серьёзные побочные эффекты, поскольку эти молекулы также участвуют в нормальном заживлении и иммунном равновесии. Новая работа задаётся вопросом, не может ли один конкретный член семейства, называемый TGF‑beta3, на самом деле помогать сердцу защищаться от неконтролируемого фиброза, а не приводить к нему.
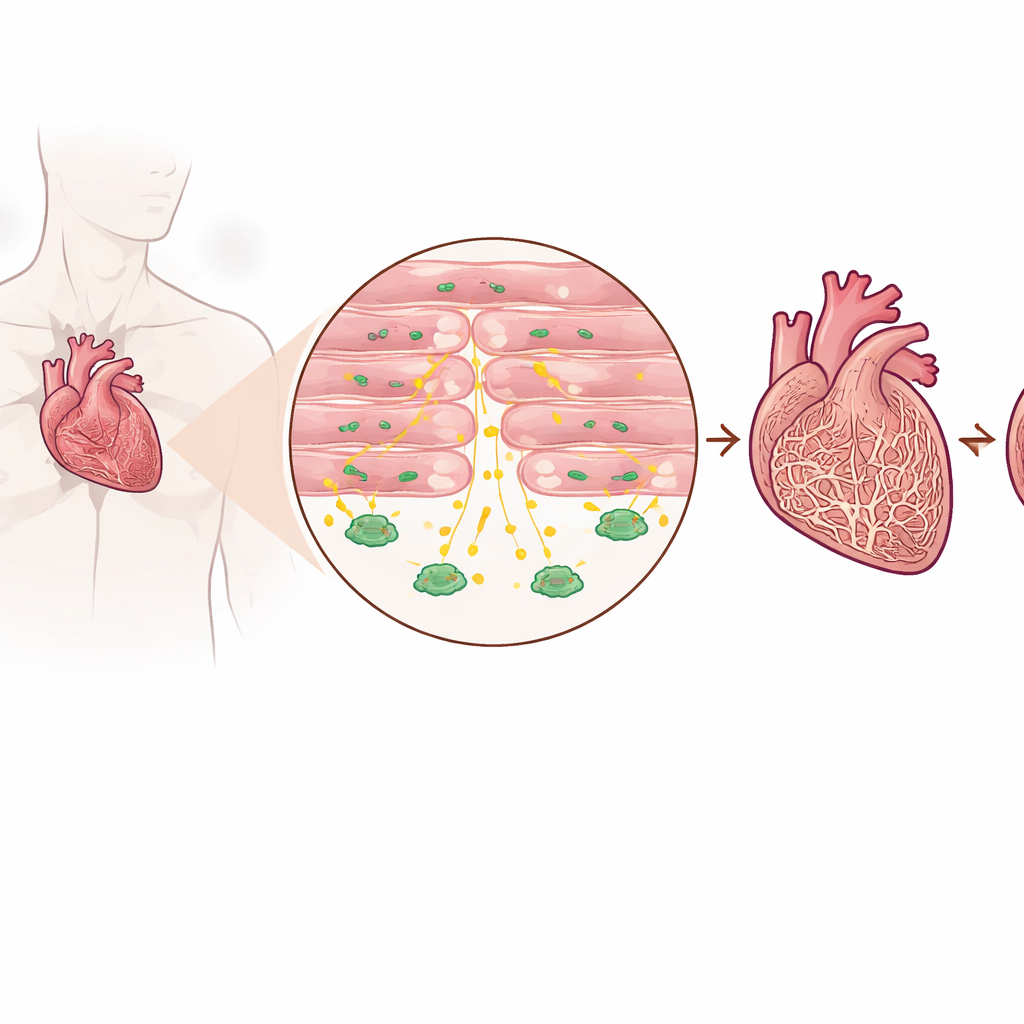
Обнаружение защитного сигнала в больных сердцах
Исследователи изучали ткань сердца у пациентов с прогрессирующей сердечной недостаточностью и у людей без неё, а также несколько мышиных моделей, имитирующих повышенную нагрузку на сердце. Они обнаружили, что уровни TGF‑beta3 последовательно были выше в поражённых сердцах по сравнению со здоровыми, как в самой ткани, так и в кровотоке. Важно, что это повышение происходило преимущественно не от привычных клеток, формирующих рубцы, а от кардиомиоцитов — мышечных клеток, которые сокращаются, чтобы перекачивать кровь. У пациентов с более высокими уровнями TGF‑beta3 в крови также, как правило, были повышены стандартные маркёры сердечной недостаточности, что свидетельствует о том, что эта молекула отражает степень стресса, испытываемого сердцем.
Выключение сигнала в кардиомиоцитах
Чтобы проверить, полезен ли этот TGF‑beta3, производимый кардиомиоцитами, команда создала мышей, в которых ген TGF‑beta3 можно было выборочно удалять только в кардиомиоцитах. В нормальных условиях в покое у этих животных сердца выглядели и функционировали почти как у их сиблинов. Но при операции, сужающей главный оттоковый сосуд сердца — стандартном способе вызвать хроническую перегрузку давлением и последующую сердечную недостаточность — различия оказались заметными. Мыши без TGF‑beta3 в кардиомиоцитах показали худшую насосную функцию, увеличение размеров сердца и значительно большее накопление коллагена между мышечными волокнами по сравнению с контрольными животными, подвергшимися тому же стрессу.
Как клетки сердца общаются с клетками, формирующими рубцы
Разбираясь в механизме, исследователи сравнили активность генов в сердцах с и без кардиомиоцитарного TGF‑beta3. Сердца, лишённые этого сигнала, показали резкое увеличение двух хорошо известных драйверов фиброза — CTGF и SERPINE1, которые преимущественно продуцируются фибробластами, клетками, ответственными за образование соединительной ткани. В клеточных культурах фибробласты, подвергнутые классическому профибротическому сигналу TGF‑beta1, включали эти фибротические гены и переходили в активированное, матриксо‑производящее состояние. Однако добавление TGF‑beta3 ослабляло эту реакцию. Фибробласты, культивированные в среде от кардиомиоцитов, лишённых TGF‑beta3, становились более активированными, чем те, что получали среду от нормальных кардиомиоцитов, подтверждая, что мышечные клетки обычно секретируют фактор, сдерживающий чрезмерную активность фибробластов.
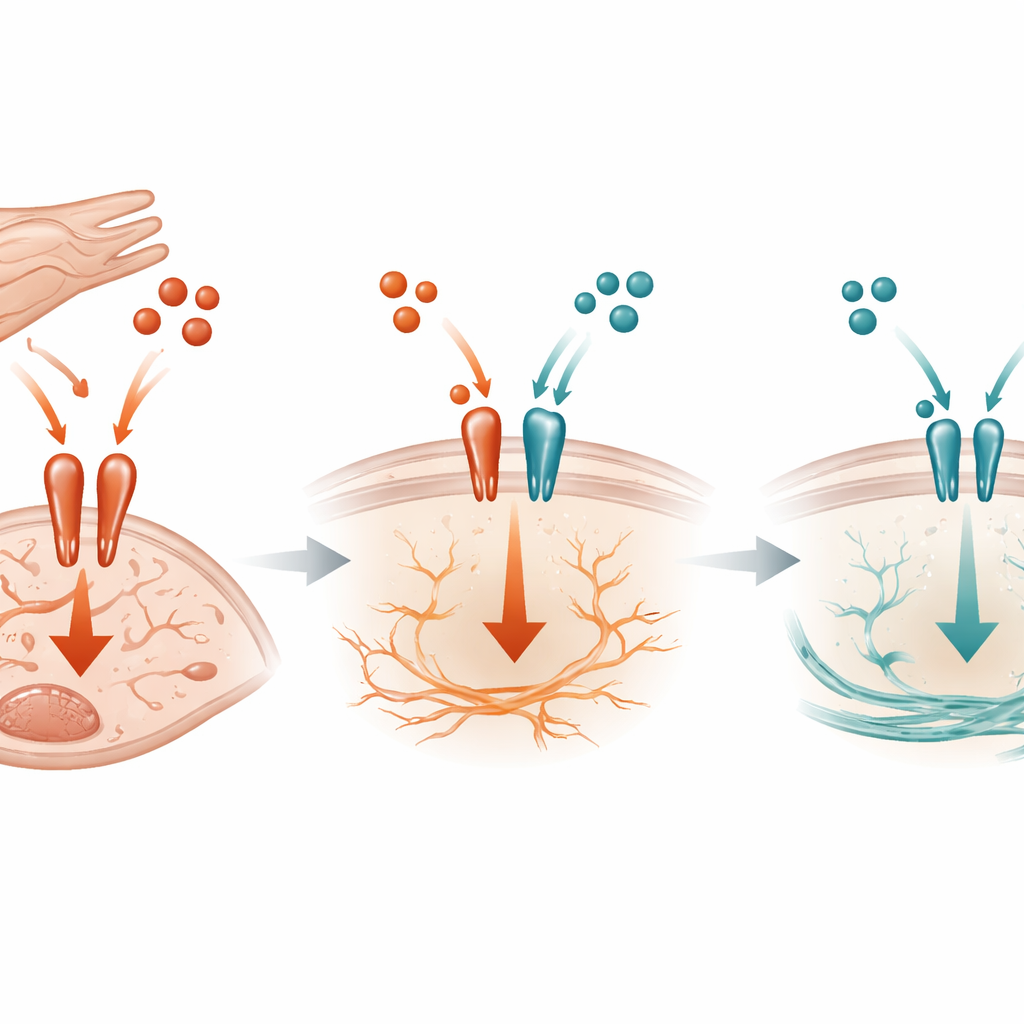
Молекулярная «перетягивание каната» на поверхности клетки
На молекулярном уровне исследование показывает, что TGF‑beta3 действует как конкурентный тормоз для более агрессивного сигнала TGF‑beta1. Обе молекулы используют один и тот же рецепторный комплекс на поверхности фибробластов для запуска внутриклеточной активности. Биохимические эксперименты показали, что TGF‑beta3 может связываться с этими рецепторами и ограничивать возможность TGF‑beta1 прикрепляться, тем самым уменьшая активацию ключевого переключателя внутри клетки — SMAD3. Когда кардиомиоцитарный TGF‑beta3 отсутствует у мышей при перегрузке давлением, активация SMAD3 возрастает, уровни CTGF и SERPINE1 повышаются, и фиброз ускоряется. Примечательно, что этот эффект не зависел от другого известного ингибитора в пути, что указывает на то, что простая конкуренция за рецептор является центральной частью защитного механизма.
Что это может значить для будущих методов лечения
Для неспециалиста ключевое послание в том, что поражённое сердце — не только жертва рубцевания; оно также пытается защититься. Кардиомиоциты выделяют TGF‑beta3 как местный, встроенный механизм защиты, который противостоит более сильным профибротическим сигналам и не даёт соединительной ткани перейти в режим гиперактивности. При отсутствии этого защитного сигнала фиброз усиливается, и функция сердца снижается быстрее. Вместо блокирования всей системы TGF‑beta будущие терапии могли бы стремиться усиливать или имитировать TGF‑beta3 конкретно в сердце, усиливая собственную способность органа ограничивать вредный фиброз при сохранении необходимого заживления в других тканях.
Цитирование: Xuan, J., Zhou, J., Huang, Y. et al. Cardiomyocyte-derived TGFB3 attenuates cardiac fibrosis and preserves cardiac function in heart failure. Sci Rep 16, 11534 (2026). https://doi.org/10.1038/s41598-026-42367-5
Ключевые слова: сердечная недостаточность, кардиальный фиброз, ТФГ-бета3, кардиомиоциты, сигнализация фибробластов