Clear Sky Science · es
La TGFβ3 derivada de los cardiomiocitos atenúa la fibrosis cardíaca y preserva la función cardíaca en la insuficiencia cardiaca
Por qué esto importa para personas con corazones débiles
La insuficiencia cardiaca es una afección frecuente y grave en la que el corazón pierde gradualmente su capacidad para bombear sangre. Un culpable principal es la cicatrización dentro del corazón, llamada fibrosis, que endurece el músculo cardíaco y empeora los síntomas. Este estudio descubre una señal protectora sorprendente producida por las propias células musculares del corazón que puede frenar este proceso de cicatrización y ayudar a conservar la función cardíaca, apuntando hacia una forma nueva y más precisa de tratar la insuficiencia cardiaca.
Una mirada más cercana a la cicatrización cardíaca
En la insuficiencia cardiaca, el corazón no se limita a “gastarse”. En cambio, su tejido se remodela: las células musculares se agrandan o mueren, y las células del tejido conectivo depositan exceso de colágeno, formando áreas rígidas similares a cicatrices. Durante años, una familia potente de moléculas señalizadoras conocida como TGF-beta se ha relacionado con esta cicatrización. Sin embargo, bloquear todas las formas de TGF-beta demostró ser un instrumento demasiado toscamente afinado, provocando efectos secundarios graves porque estas moléculas también ayudan en la reparación normal y en el equilibrio inmunitario. El nuevo trabajo plantea si un miembro particular de esta familia, llamado TGF-beta3, podría en realidad ayudar al corazón a defenderse contra la fibrosis descontrolada en lugar de impulsarla.
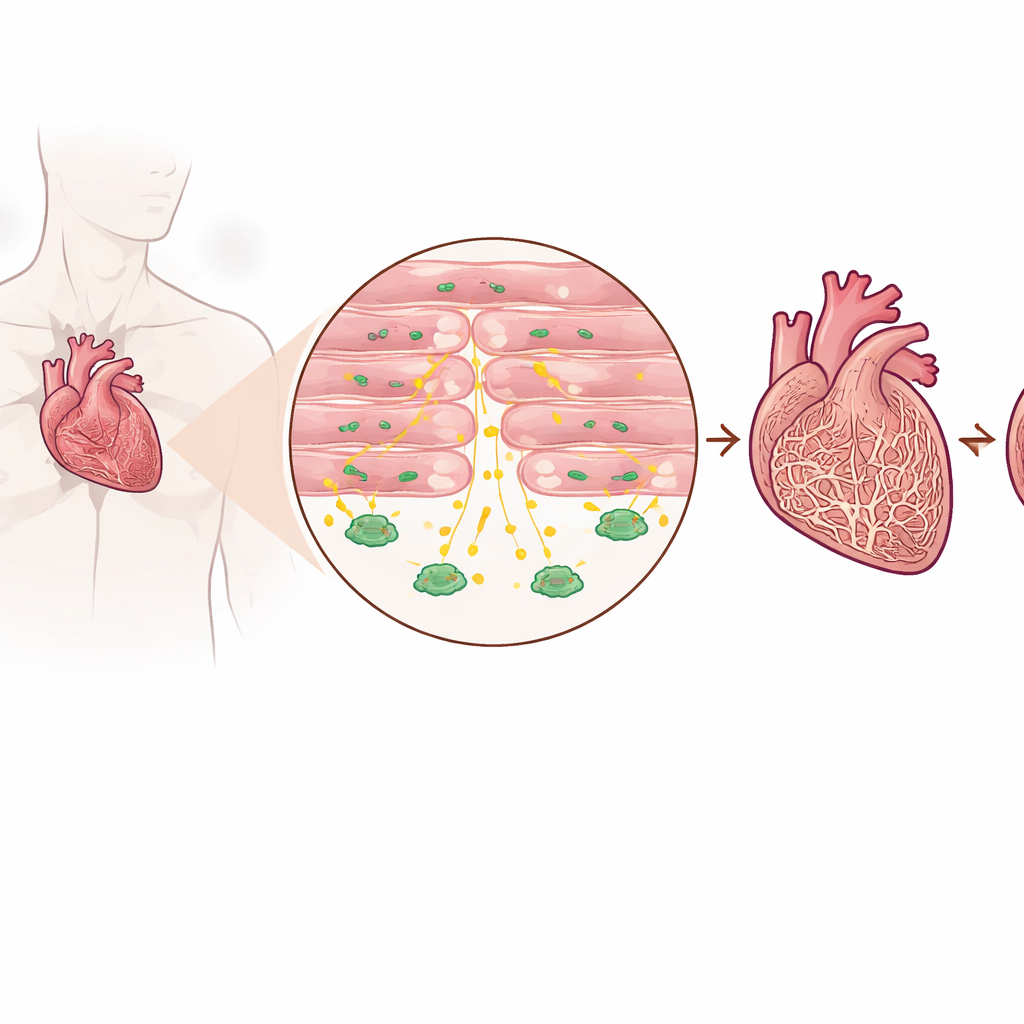
Detectando una señal protectora en corazones enfermos
Los investigadores examinaron tejido cardíaco de pacientes con insuficiencia cardiaca avanzada y de personas sin insuficiencia, así como varios modelos murinos que imitan la sobrecarga de presión en el corazón. Encontraron que los niveles de TGF-beta3 eran consistentemente más altos en corazones fallidos que en los sanos, tanto en el propio tejido como en el torrente sanguíneo. De manera importante, este aumento no provenía principalmente de las células formadoras de cicatriz habituales, sino de los cardiomiocitos: las células musculares que realmente se contraen para bombear sangre. Los pacientes con niveles sanguíneos más altos de TGF-beta3 también tendían a tener niveles más elevados de un marcador estándar de insuficiencia cardiaca, lo que sugiere que esta molécula refleja la intensidad del estrés que soporta el corazón.
Apagar la señal en las células musculares del corazón
Para probar si la TGF-beta3 producida por los cardiomiocitos era beneficiosa o perjudicial, el equipo diseñó ratones en los que el gen de la TGF-beta3 podía eliminarse selectivamente solo en las células musculares del corazón. En condiciones normales y en reposo, estos ratones tenían corazones que se veían y funcionaban de forma similar a los de sus compañeros. Pero cuando los animales fueron sometidos a un procedimiento que constriñe la arteria principal que sale del corazón—una forma estándar de inducir sobrecarga crónica de presión y eventual insuficiencia—las diferencias se hicieron notables. Los ratones sin TGF-beta3 en sus cardiomiocitos desarrollaron una función de bombeo peor, corazones más grandes y mucha más acumulación de colágeno entre las fibras musculares que los animales de control expuestos al mismo estrés.
Cómo las células musculares del corazón hablan con las formadoras de cicatriz
Profundizando en el mecanismo, los investigadores compararon la actividad génica en corazones con y sin TGF-beta3 de cardiomiocitos. Los corazones que carecían de esta señal mostraron un aumento marcado en dos impulsores bien conocidos de la fibrosis, CTGF y SERPINE1, que se producen principalmente por los fibroblastos, las células responsables de fabricar tejido conectivo. En experimentos en cultivo celular, cuando los fibroblastos fueron expuestos a la señal clásica de cicatrización TGF-beta1, activaron estos genes fibróticos y adoptaron un estado activado productor de matriz. Añadir TGF-beta3 a la mezcla, sin embargo, atenuó esa respuesta. Los fibroblastos bañados en el fluido procedente de cardiomiocitos deficientes en TGF-beta3 se activaron más que los expuestos al fluido de cardiomiocitos normales, confirmando que las células musculares normalmente secretan un factor que frena la sobreactividad de los fibroblastos.
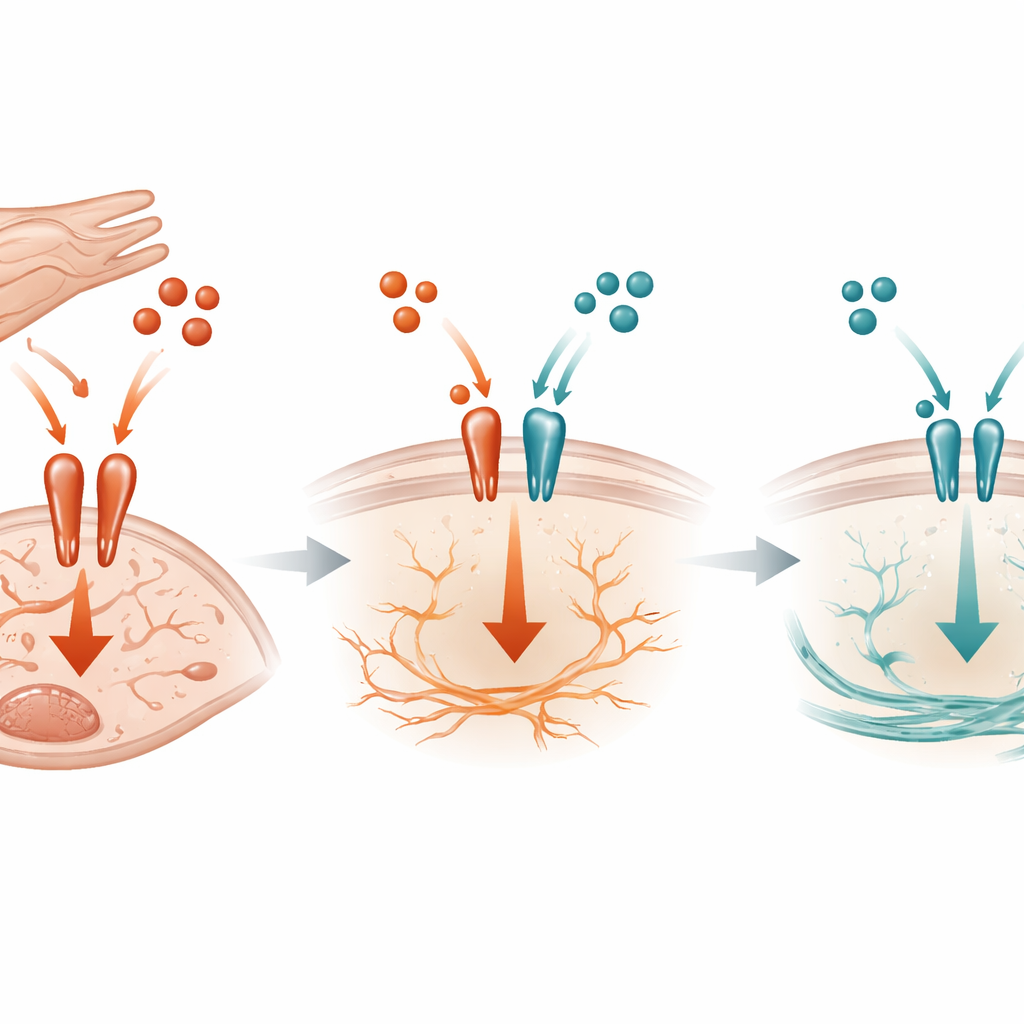
Una lucha molecular en la superficie celular
A nivel molecular, el estudio muestra que la TGF-beta3 actúa como un freno competitivo frente a la señal más agresiva TGF-beta1. Ambas moléculas usan el mismo complejo receptor en la superficie de los fibroblastos para desencadenar actividad dentro de la célula. Experimentos bioquímicos revelaron que la TGF-beta3 puede unirse a estos receptores y limitar cuánto puede adherirse la TGF-beta1, reduciendo así la activación de un interruptor clave dentro de la célula llamado SMAD3. Cuando la TGF-beta3 derivada de los cardiomiocitos está ausente en ratones bajo sobrecarga de presión, la activación de SMAD3 aumenta, CTGF y SERPINE1 se incrementan y la fibrosis se acelera. Cabe destacar que este efecto no dependió de otro inhibidor conocido dentro de la vía, lo que sugiere que la simple competencia por el receptor es una parte central del mecanismo protector.
Qué podría significar esto para tratamientos futuros
Para un público no especializado, el mensaje clave es que el corazón fallido no es solo víctima de la cicatrización; también intenta protegerse. Las células musculares del corazón liberan TGF-beta3 como una defensa local incorporada que compite contra señales pro-cicatrización más fuertes y evita que el tejido conectivo entre en sobreproducción. Cuando esta señal protectora falta, la fibrosis empeora y la función cardíaca declina más rápido. En lugar de bloquear todo el sistema TGF-beta, las futuras terapias podrían buscar potenciar o imitar específicamente la TGF-beta3 en el corazón, reforzando la capacidad del órgano para limitar la cicatrización dañina mientras se preserva la reparación necesaria en otros tejidos.
Cita: Xuan, J., Zhou, J., Huang, Y. et al. Cardiomyocyte-derived TGFB3 attenuates cardiac fibrosis and preserves cardiac function in heart failure. Sci Rep 16, 11534 (2026). https://doi.org/10.1038/s41598-026-42367-5
Palabras clave: insuficiencia cardiaca, fibrosis cardíaca, TGF-beta3, cardiomiocitos, señalización de fibroblastos