Clear Sky Science · sv
scTWAS: en kraftfull statistisk ram för transcriptome-wide association-studier i enskilda celler
Varför att studera enskilda celler kan förändra medicinen
De flesta genetiska studier av sjukdom fungerar som att lyssna på en folkmassa: de uppfattar ett samlat dån men missar vad varje individ säger. Denna artikel visar hur vi kan ställa in öronen på enskilda röster. Författarna presenterar scTWAS, ett nytt sätt att koppla DNA‑variationer till sjukdom genom att studera genaktivitet i specifika celltyper och ännu finare cellsubtyper med hjälp av single-cell RNA‑sekvenseringsdata. Den skarpare bilden avslöjar vilka exakta celler — och vilka gener i dem — som driver tillstånd som blodsjukdomar, autoimmuna sjukdomar och Alzheimers sjukdom.
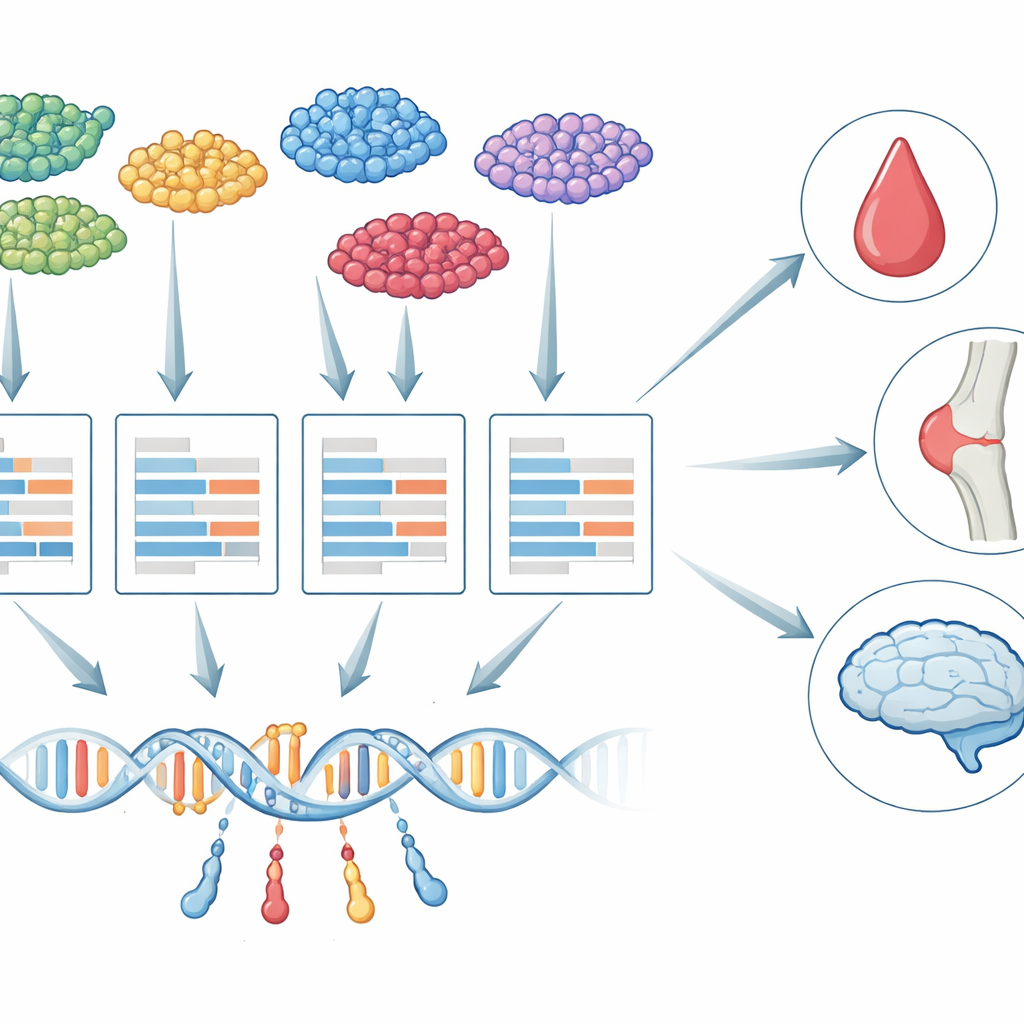
Från bulkvävnad till enskilda celler
I mer än ett decennium har forskare använt en strategi kallad transcriptome-wide association studies (TWAS) för att koppla genetiska varianter till sjukdomar. TWAS fungerar i två steg: först lär den sig hur DNA‑förändringar påverkar genaktivitet, sedan testar den om den genetiskt predikterade aktiviteten av varje gen är kopplad till en egenskap, såsom trombocytantal eller demensrisk. Hittills har nästan all TWAS‑forskning förlitat sig på "bulk"‑vävnadsprover, där RNA från många celltyper blandas. Denna blandning döljer viktiga skillnader: den genetiska kontrollen av en gen i en mikroglia i hjärnan kan till exempel vara mycket annorlunda än samma gen i en neuron, och bara vissa av dessa celler kan verkligen vara avgörande för en given sjukdom.
Problemet med brusiga single-cell‑data
Nya populationstäckande single-cell RNA‑sekvenseringsmetoder gör det nu möjligt att mäta tusentals individuella celler per person, över många personer. Men dessa data är röriga: räkningarna är glesa (många nollor), starkt påverkade av tekniska egenheter i experimentet och varierar stort från cell till cell även när biologin är densamma. Tidigare försök att använda single-cell‑data i TWAS använde ad hoc‑normaliseringstricks hämtade från bulk‑RNA‑metoder i hopp om att tygla bruset. Författarna visar att sådana genvägar kan förvränga de verkliga genetiska effekterna på genaktivitet, vilket leder till svagare prediktioner och färre upptäckta gen–sjukdom‑kopplingar, särskilt i sällsynta eller svårfångade celltyper.
Hur scTWAS renar signalen
scTWAS tar sig an dessa utmaningar genom att uttryckligen skilja biologi från mätfel. Först aggregerar den single-cell‑räkningar inom varje person och celltyp till en "pseudo‑bulk"‑profil, vilket minskar gleshet samtidigt som celltypidentiteten bevaras. Sedan använder den en tvålagers statistisk modell: ett lager beskriver hur DNA‑varianter och grundläggande egenskaper som ålder påverkar en persons verkliga underliggande genaktivitet i en given celltyp; det andra lagret modellerar hur sekvenseringsmaskinen omvandlar den aktiviteten till brusiga räkningar, inklusive effekten av varierande sekvenseringsdjup. Genom att passa denna modell med en specialiserad viktad regressionsalgoritm nedväger scTWAS de mest brusiga proverna och uppskattar den genetiskt reglerade uttrycksnivån för varje gen i varje celltyp mer noggrant.
Hitta sjukdomsgener där de verkligen verkar
När dessa celltypsspecifika prediktionsmodeller är tränade, pluggar scTWAS in dem i stora genome‑wide association‑studier för att testa gen–egenskapskopplingar. I simulerade data som efterliknar verkliga single-cell‑experiment presterade scTWAS konsekvent bättre än befintliga metoder både i prediktionsnoggrannhet och i statistisk styrka, med särskilt stora vinster för sällsynta celltyper där data är knappa. När ramverket tillämpades på immunceller visar författarna att scTWAS identifierar avsevärt fler gener kopplade till 29 blodegenskaper samt till reumatoid artrit, lupus och astma. Många av dessa signaler lyfter fram särskilda immuncellstyper — såsom specifika monocyter eller T‑cellsundergrupper — som huvudscenen där vissa gener påverkar sjukdomsrisken, och vissa associationer missades helt i bulk‑blodanalyser.
En titt i hjärnans cellsubtyper vid Alzheimers
scTWAS blir ännu mer avslöjande i hjärnan. Med single‑nucleus‑data från hundratals donerade mänskliga hjärnor byggde författarna prediktionsmodeller för sex stora hjärncellstyper och 75 finare subtyper. De kombinerade dessa med Alzheimers genetiska data för att kartlägga var, på cellulär nivå, riskgener sannolikt verkar. Vissa gener dyker upp i många celltyper, vilket antyder breda roller i hjärnan, medan andra är slående specifika. Till exempel visar en känd riskgen, MS4A6A, en stark association endast i en sjukdomsassocierad mikroglial subtyp kopplad till lipidhantering, och PPP1R37 är associerad endast i en inflammatorisk mikroglial subtyp nära den välkända APOE‑riskregionen. Dessa mönster pekar på distinkta mikrogliala tillstånd som nyckelaktörer i hur vissa genetiska varianter driver Alzheimersrisk.

Vad detta betyder för framtida terapier
För en icke‑specialist är huvudbudskapet att var en gen verkar kan vara lika viktigt som vad genen gör. Genom att flytta TWAS från blandade vävnader ner till precisa celltyper och subtyper, och genom att noggrant modellera egenheterna i single‑cell‑mätningar, avslöjar scTWAS gen–sjukdom‑kopplingar som tidigare var osynliga. Denna skarpare karta hjälper forskare att rikta in sig på exakt vilka cellpopulationer och signalvägar som bör vara mål för nya läkemedel eller interventioner, från blodbildning och immunförsvar till hjärnans immunceller i Alzheimers sjukdom.
Citering: Lin, Z., Su, C. scTWAS: a powerful statistical framework for single-cell transcriptome-wide association studies. Nat Commun 17, 3853 (2026). https://doi.org/10.1038/s41467-026-70374-7
Nyckelord: single-cell genomik, kartläggning av genetisk risk, immunceller, Alzheimers sjukdom, statistisk genetik