Clear Sky Science · es
scTWAS: un potente marco estadístico para estudios de asociación a nivel del transcriptoma en células individuales
Por qué mirar a las células individuales puede cambiar la medicina
La mayoría de los estudios genéticos de enfermedades funcionan como escuchar a una multitud: captan un rugido general pero no lo que dice cada persona. Este artículo muestra cómo afinar el oído para oír voces individuales. Los autores presentan scTWAS, una nueva forma de conectar las diferencias en el ADN con la enfermedad mediante la observación de la actividad génica en tipos celulares específicos e incluso en subtipos más finos, usando datos de secuenciación de ARN de célula única. Esa visión más nítida revela qué células concretas —y qué genes dentro de ellas— están impulsando afecciones como trastornos sanguíneos, enfermedades autoinmunes y la enfermedad de Alzheimer.
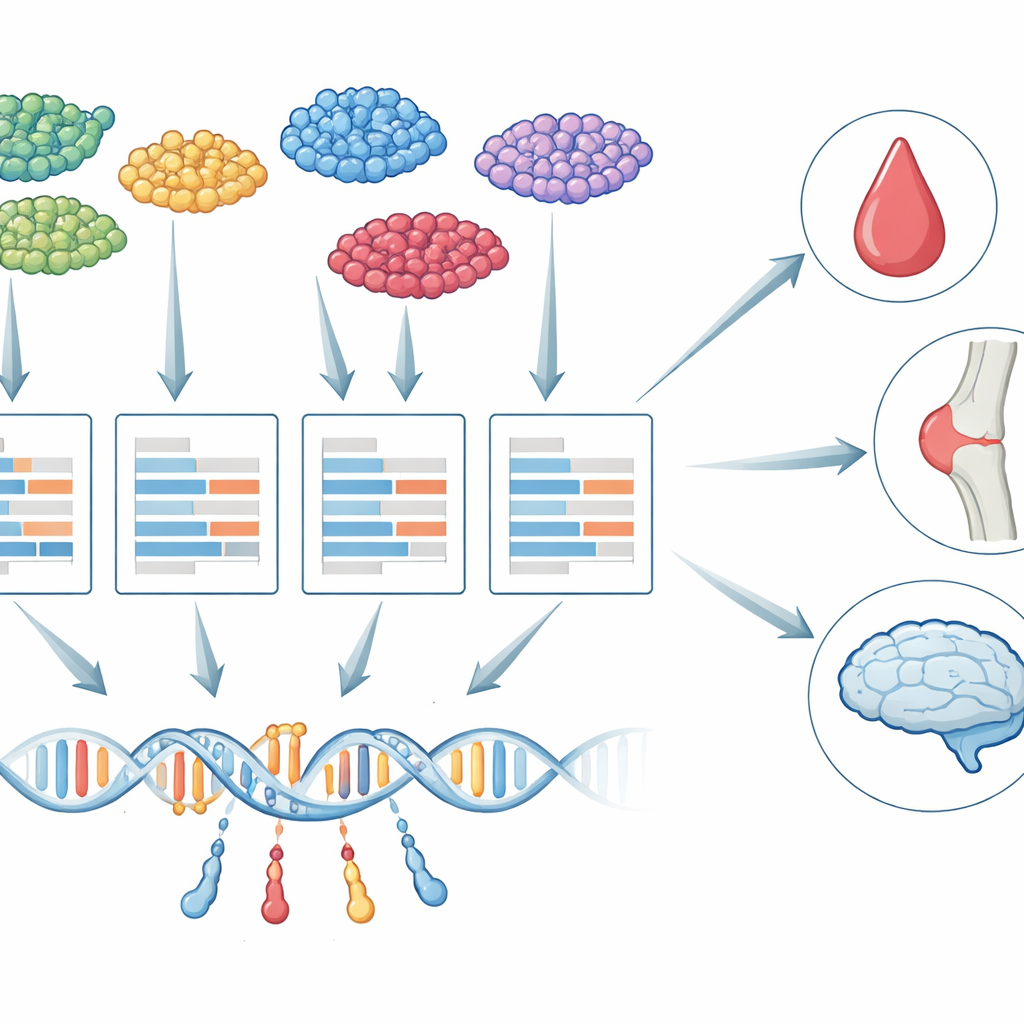
De tejido agregado a células individuales
Durante más de una década, los investigadores han utilizado una estrategia llamada estudios de asociación a nivel del transcriptoma (TWAS) para vincular variantes genéticas con enfermedades. TWAS funciona en dos pasos: primero aprende cómo los cambios en el ADN influyen en la actividad génica, y luego prueba si la actividad de cada gen, predicha por la genética, está relacionada con un rasgo como el recuento de plaquetas o el riesgo de demencia. Hasta ahora, casi todo el trabajo de TWAS se ha basado en muestras de tejido “agregado” (bulk), donde el ARN de muchos tipos celulares se mezcla. Esa mezcla oculta diferencias importantes: el control genético de un gen en una microglía del cerebro, por ejemplo, puede ser muy distinto del mismo gen en una neurona, y solo algunos de esos tipos celulares pueden ser relevantes para una enfermedad determinada.
El problema de los datos ruidosos de célula única
La nueva secuenciación de ARN a escala poblacional por célula única ahora permite medir miles de células individuales por persona, en muchas personas. Pero estos datos son desordenados: los recuentos son escasos (muchos ceros), están fuertemente influenciados por peculiaridades técnicas del experimento y varían mucho de célula a célula incluso cuando la biología es la misma. Intentos previos de integrar datos de célula única en TWAS usaron trucos de normalización ad hoc tomados de métodos de RNA bulk, esperando que esos pasos domaran el ruido. Los autores muestran que esos atajos pueden distorsionar los verdaderos efectos genéticos sobre la actividad génica, llevando a predicciones más débiles y a menos enlaces gen–enfermedad descubiertos, especialmente en tipos celulares raros o difíciles.
Cómo scTWAS limpia la señal
scTWAS afronta estos desafíos separando explícitamente la biología del error de medida. Primero, agrega los recuentos de célula única dentro de cada persona y tipo celular en un perfil de “pseudo-bulk”, reduciendo la escasez a la vez que preserva la identidad del tipo celular. Luego utiliza un modelo estadístico de dos capas: una capa describe cómo las variantes del ADN y características básicas como la edad influyen en la verdadera actividad génica subyacente de una persona en un determinado tipo celular; la otra capa modela cómo la máquina de secuenciación transforma esa actividad en recuentos ruidosos, incluida la influencia de la profundidad variable de secuenciación. Al ajustar este modelo con un algoritmo de regresión ponderada especializado, scTWAS resta peso a las muestras más ruidosas y estima con mayor precisión la expresión regulada por la genética para cada gen en cada tipo celular.
Encontrar genes de enfermedad donde realmente actúan
Una vez entrenados estos modelos de predicción específicos por tipo celular, scTWAS los aplica a grandes estudios de asociación del genoma para probar enlaces gen–rasgo. En datos simulados que imitan experimentos reales de célula única, scTWAS superó de forma consistente a los métodos existentes tanto en precisión de predicción como en potencia, con ganancias especialmente grandes en tipos celulares raros donde los datos son más escasos. Al aplicar el marco a células inmunitarias, los autores muestran que scTWAS identifica muchos más genes asociados con 29 rasgos sanguíneos y con artritis reumatoide, lupus y asma. Muchas de estas señales resaltan tipos celulares inmunitarios concretos —como subconjuntos específicos de monocitos o células T— como el escenario principal en el que ciertos genes afectan el riesgo de enfermedad, y algunas asociaciones fueron totalmente pasadas por alto en análisis de sangre en bulk.
Escudriñando subtipos celulares del cerebro en Alzheimer
scTWAS resulta aún más revelador en el cerebro. Usando datos de núcleos individuales de cientos de cerebros donados, los autores construyeron modelos predictivos para seis tipos celulares principales del cerebro y 75 subtipos más finos. Luego combinaron estos modelos con datos genéticos de la enfermedad de Alzheimer para mapear dónde, a resolución celular, es más probable que actúen los genes de riesgo. Algunos genes aparecen en muchos tipos celulares, lo que sugiere roles amplios en el cerebro, mientras que otros son notablemente específicos. Por ejemplo, un gen de riesgo conocido, MS4A6A, muestra una fuerte asociación solo en un subtipo microglial asociado a la enfermedad vinculado al manejo de lípidos, y PPP1R37 se asocia únicamente en un subtipo microglial inflamatorio cerca de la conocida región de riesgo APOE. Estos patrones señalan a estados microgliales distintos como actores clave en cómo ciertas variantes genéticas impulsan el riesgo de Alzheimer.

Qué significa esto para futuras terapias
Para un lector no especialista, el mensaje principal es que dónde actúa un gen puede ser tan importante como lo que hace ese gen. Al trasladar TWAS de tejidos mezclados a tipos celulares y subtipos precisos, y al modelar con cuidado las peculiaridades de las mediciones de célula única, scTWAS descubre conexiones gen–enfermedad que antes eran invisibles. Este mapa más nítido ayuda a los investigadores a centrar su atención en las poblaciones celulares y vías exactas que deberían ser el objetivo de nuevos fármacos o intervenciones, desde la formación de la sangre y la inmunidad hasta las células inmunitarias del cerebro en la enfermedad de Alzheimer.
Cita: Lin, Z., Su, C. scTWAS: a powerful statistical framework for single-cell transcriptome-wide association studies. Nat Commun 17, 3853 (2026). https://doi.org/10.1038/s41467-026-70374-7
Palabras clave: genómica de célula única, mapeo del riesgo genético, células inmunitarias, enfermedad de Alzheimer, genética estadística