Clear Sky Science · pt
scTWAS: uma poderosa estrutura estatística para estudos de associação transcriptoma-ampla em célula única
Por que olhar para células individuais pode mudar a medicina
A maioria dos estudos genéticos de doenças funciona como ouvir uma multidão: percebe-se um rugido geral, mas perde-se o que cada pessoa está dizendo. Este artigo mostra como afinar nossos ouvidos para vozes individuais. Os autores apresentam o scTWAS, um novo método para conectar variações no DNA a doenças examinando a atividade gênica em tipos celulares específicos e até subtipos celulares mais finos, usando dados de RNA-seq de célula única. Essa visão mais precisa revela quais células exatas — e quais genes dentro delas — estão impulsionando condições como desordens sanguíneas, doenças autoimunes e a doença de Alzheimer.
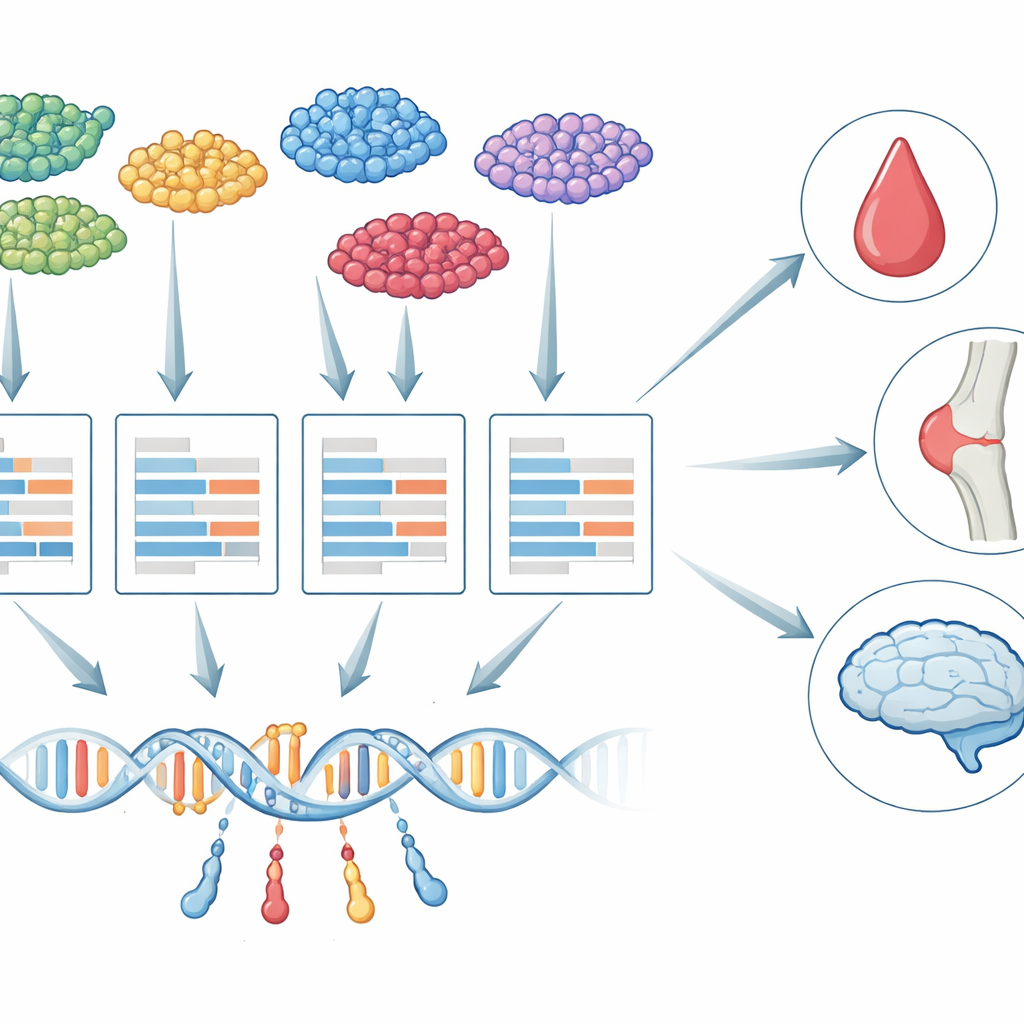
Do tecido agregado às células individuais
Por mais de uma década, pesquisadores têm usado uma estratégia chamada estudos de associação transcriptoma-ampla (TWAS) para ligar variantes genéticas a doenças. O TWAS funciona em dois passos: primeiro aprende como mudanças no DNA influenciam a atividade gênica, depois testa se a atividade de cada gene prevista geneticamente está ligada a um traço, como contagem de plaquetas ou risco de demência. Até agora, quase todos os trabalhos de TWAS dependeram de amostras de tecido “agregado” (bulk), onde o RNA de muitos tipos celulares é misturado. Essa mistura oculta diferenças importantes: o controle genético de um gene em um micróglia no cérebro, por exemplo, pode ser muito diferente do mesmo gene em um neurônio, e apenas alguns desses tipos celulares podem realmente importar para uma dada doença.
O problema dos dados ruidosos de célula única
Novas coletas populacionais de RNA-seq de célula única agora permitem medir milhares de células individuais por pessoa, em muitas pessoas. Mas esses dados são bagunçados: as contagens são esparsas (muitos zeros), fortemente afetadas por peculiaridades técnicas do experimento e variam muito de célula para célula mesmo quando a biologia é a mesma. Tentativas anteriores de inserir dados de célula única no TWAS usaram truques de normalização ad hoc emprestados de métodos de bulk, esperando que essas etapas domassem o ruído. Os autores mostram que esses atalhos podem distorcer os verdadeiros efeitos genéticos sobre a atividade gênica, levando a previsões mais fracas e a menos associações gene–doença detectadas, especialmente em tipos celulares raros ou difíceis.
Como o scTWAS limpa o sinal
O scTWAS enfrenta esses desafios separando explicitamente a biologia do erro de medição. Primeiro, agrega as contagens de célula única dentro de cada pessoa e tipo celular em um perfil “pseudo-bulk”, reduzindo a esparsidade ao mesmo tempo em que preserva a identidade do tipo celular. Em seguida, usa um modelo estatístico de duas camadas: uma camada descreve como variantes do DNA e características básicas como idade influenciam a verdadeira atividade gênica subjacente de uma pessoa em um dado tipo celular; a outra camada modela como a máquina de sequenciamento transforma essa atividade em contagens ruidosas, incluindo o efeito da profundidade de sequenciamento variável. Ao ajustar esse modelo com um algoritmo de regressão ponderada especializado, o scTWAS reduz o peso das amostras mais ruidosas e estima com mais precisão a expressão regulada geneticamente para cada gene em cada tipo celular.
Encontrando genes de doença onde eles realmente atuam
Uma vez que esses modelos de predição específicos por tipo celular são treinados, o scTWAS os integra em grandes estudos de associação genômica para testar ligações gene–traço. Em dados simulados que imitam experimentos reais de célula única, o scTWAS superou consistentemente métodos existentes tanto em precisão de previsão quanto em potência, com ganhos especialmente grandes para tipos celulares raros, onde os dados são mais escassos. Aplicando a estrutura a células imunes, os autores mostram que o scTWAS identifica substancialmente mais genes associados a 29 traços sanguíneos e a artrite reumatoide, lúpus e asma. Muitos desses sinais destacam tipos celulares imunes particulares — como subpopulações específicas de monócitos ou células T — como o principal palco no qual certos genes afetam o risco de doença, e algumas associações foram totalmente perdidas por análises em sangue agregado.
Observando subtipos de células cerebrais no Alzheimer
O scTWAS torna-se ainda mais revelador no cérebro. Usando dados de núcleos únicos de centenas de cérebros humanos doados, os autores construíram modelos de predição para seis grandes tipos celulares cerebrais e 75 subtipos mais finos. Em seguida, combinaram esses modelos com dados genéticos da doença de Alzheimer para mapear onde, em resolução celular, os genes de risco provavelmente atuam. Alguns genes aparecem em muitos tipos celulares, sugerindo papéis amplos no cérebro, enquanto outros são notavelmente específicos. Por exemplo, um gene de risco conhecido, MS4A6A, mostra uma forte associação apenas em um subtipo de micróglia associado à doença e ligado ao manejo de lipídios, e PPP1R37 está associado apenas em um subtipo microgial inflamatório perto da bem conhecida região de risco APOE. Esses padrões apontam para estados microgiais distintos como atores-chave em como certas variantes genéticas impulsionam o risco de Alzheimer.

O que isso significa para terapias futuras
Para um não-especialista, a mensagem principal é que onde um gene atua pode ser tão importante quanto o que o gene faz. Ao mover o TWAS de tecidos misturados para tipos e subtipos celulares precisos, e ao modelar cuidadosamente as peculiaridades das medições de célula única, o scTWAS revela conexões gene–doença que antes eram invisíveis. Esse mapa mais nítido ajuda pesquisadores a focalizar as populações celulares e vias exatas que devem ser alvo para novos fármacos ou intervenções, desde a formação do sangue e imunidade até as células imunes do cérebro na doença de Alzheimer.
Citação: Lin, Z., Su, C. scTWAS: a powerful statistical framework for single-cell transcriptome-wide association studies. Nat Commun 17, 3853 (2026). https://doi.org/10.1038/s41467-026-70374-7
Palavras-chave: genômica de célula única, mapeamento de risco genético, células imunes, doença de Alzheimer, genética estatística