Clear Sky Science · fr
scTWAS : un puissant cadre statistique pour les études d’association à l’échelle du transcriptome en cellules uniques
Pourquoi regarder les cellules individuelles peut changer la médecine
La plupart des études génétiques sur les maladies fonctionnent comme écouter une foule : on entend un grondement global mais on manque ce que dit chaque individu. Cet article montre comment accorder notre oreille aux voix individuelles. Les auteurs présentent scTWAS, une nouvelle méthode pour relier les différences d’ADN aux maladies en observant l’activité génique dans des types cellulaires spécifiques et même des sous-types cellulaires plus fins, à partir de données de séquençage d’ARN en cellule unique. Cette vue plus précise révèle quelles cellules exactes — et quels gènes en leur sein — sont à l’origine de pathologies telles que les troubles sanguins, les maladies auto-immunes et la maladie d’Alzheimer.
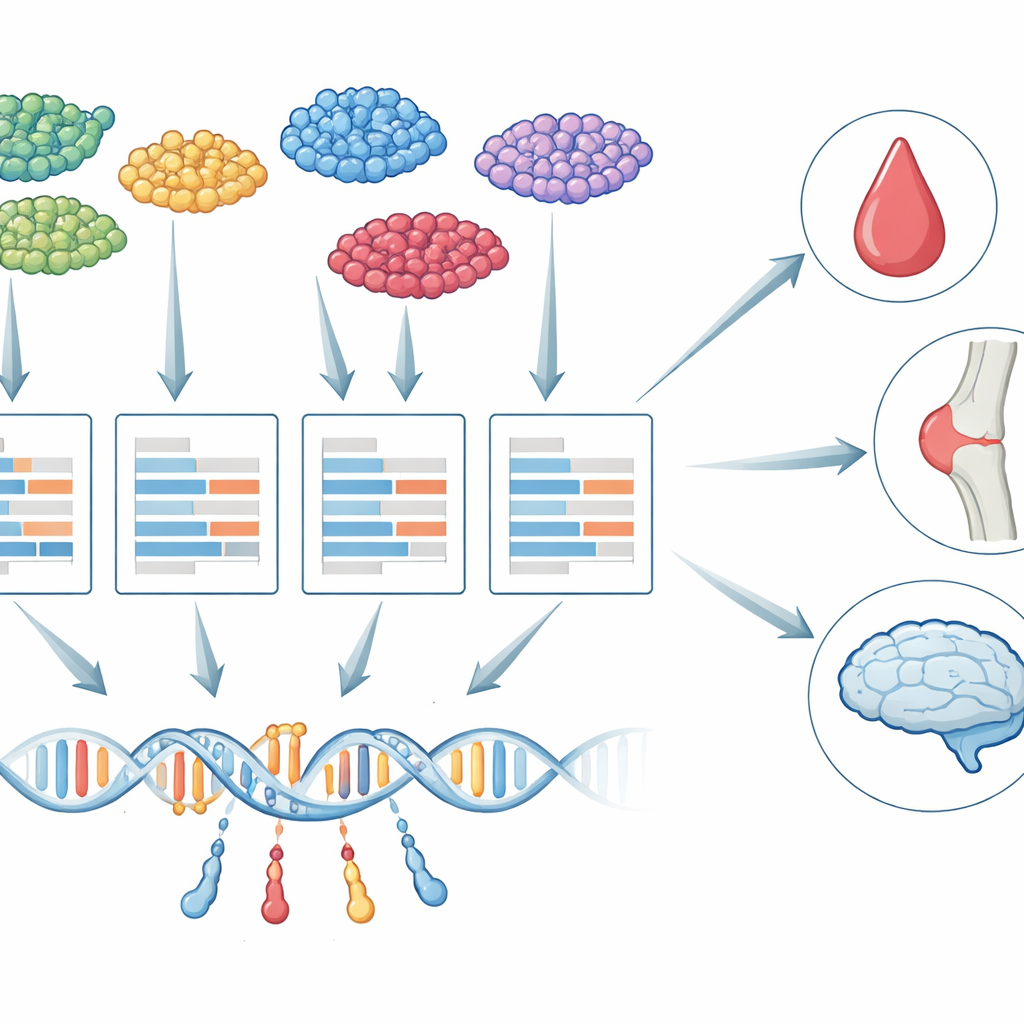
Des tissus en masse aux cellules uniques
Pendant plus d’une décennie, les chercheurs ont utilisé une stratégie appelée études d’association à l’échelle du transcriptome (TWAS) pour relier les variants génétiques aux maladies. TWAS fonctionne en deux étapes : d’abord elle apprend comment les variations d’ADN influencent l’activité des gènes, puis elle teste si l’activité prédite génétiquement de chaque gène est liée à un trait comme le nombre de plaquettes ou le risque de démence. Jusqu’à présent, presque tous les travaux TWAS se sont appuyés sur des échantillons de tissu « en masse », où l’ARN de nombreux types cellulaires est mélangé. Ce mélange masque des différences importantes : le contrôle génétique d’un gène dans une microglie du cerveau, par exemple, peut être très différent de ce même gène dans un neurone, et seules certaines de ces cellules peuvent réellement importer pour une maladie donnée.
Le problème des données en cellule unique bruyantes
Les nouvelles enquêtes à l’échelle de la population utilisant le séquençage d’ARN en cellule unique permettent désormais de mesurer des milliers de cellules individuelles par personne, sur de nombreuses personnes. Mais ces données sont bruitées : les comptes sont clairsemés (beaucoup de zéros), fortement affectés par des particularités techniques de l’expérience, et varient fortement d’une cellule à l’autre même lorsque la biologie est identique. Les tentatives antérieures d’introduire des données en cellule unique dans TWAS utilisaient des astuces de normalisation ad hoc empruntées aux méthodes sur tissu en masse, en espérant que ces étapes dompteraient le bruit. Les auteurs montrent que de tels raccourcis peuvent déformer les effets génétiques réels sur l’activité génique, conduisant à des prédictions moins précises et à moins de liens gène–maladie découverts, en particulier pour les types cellulaires rares ou difficiles.
Comment scTWAS nettoie le signal
scTWAS relève ces défis en séparant explicitement la biologie de l’erreur de mesure. D’abord, il agrège les comptes en cellule unique pour chaque personne et type cellulaire en un profil « pseudo-masse », réduisant la clairseméité tout en préservant l’identité du type cellulaire. Ensuite, il utilise un modèle statistique à deux couches : une couche décrit comment les variants d’ADN et des caractéristiques basiques comme l’âge influencent l’activité génique véritable et sous-jacente d’une personne dans un type cellulaire donné ; l’autre couche modélise comment la machine de séquençage transforme cette activité en comptes bruités, incluant l’effet d’une profondeur de séquençage variable. En ajustant ce modèle avec un algorithme de régression pondérée spécialisé, scTWAS sous-pondère les échantillons les plus bruités et estime plus précisément l’expression régulée génétiquement de chaque gène dans chaque type cellulaire.
Identifier les gènes de maladie là où ils agissent réellement
Une fois ces modèles de prédiction spécifiques aux types cellulaires entraînés, scTWAS les intègre dans de larges études d’association à l’échelle du génome pour tester les liens gène–phénotype. Dans des données simulées imitant de véritables expériences en cellule unique, scTWAS surpasse systématiquement les méthodes existantes tant en précision de prédiction qu’en puissance, avec des gains particulièrement importants pour les types cellulaires rares où les données sont les plus ténues. En appliquant le cadre aux cellules immunitaires, les auteurs montrent que scTWAS identifie nettement plus de gènes associés à 29 traits sanguins ainsi qu’à la polyarthrite rhumatoïde, au lupus et à l’asthme. Beaucoup de ces signaux mettent en évidence des types cellulaires immunitaires particuliers — comme des sous-ensembles spécifiques de monocytes ou de cellules T — comme la scène principale où certains gènes influencent le risque de maladie, et certaines associations étaient totalement invisibles dans les analyses sur sang en masse.
Explorer les sous-types cellulaires du cerveau dans la maladie d’Alzheimer
scTWAS devient encore plus révélateur dans le cerveau. En utilisant des données de noyaux uniques provenant de centaines de cerveaux humains donnés, les auteurs ont construit des modèles de prédiction pour six grands types cellulaires cérébraux et 75 sous-types plus fins. Ils ont ensuite combiné ces modèles avec des données génétiques sur la maladie d’Alzheimer pour cartographier, à résolution cellulaire, où les gènes de risque agissent probablement. Certains gènes apparaissent dans de nombreux types cellulaires, suggérant des rôles larges dans le cerveau, tandis que d’autres sont frappant de spécificité. Par exemple, un gène de risque connu, MS4A6A, montre une forte association uniquement dans un sous-type microglial associé à la maladie et lié au métabolisme des lipides, et PPP1R37 est associé uniquement dans un sous-type microglial inflammatoire proche de la région de risque bien connue APOE. Ces motifs indiquent que des états microgliaux distincts sont des acteurs clés de la façon dont certains variants génétiques augmentent le risque d’Alzheimer.

Ce que cela signifie pour les thérapies futures
Pour un non-spécialiste, le message principal est que le lieu d’action d’un gène peut être aussi important que la fonction du gène. En faisant passer TWAS de tissus mélangés à des types et sous-types cellulaires précis, et en modélisant soigneusement les particularités des mesures en cellule unique, scTWAS révèle des liens gène–maladie auparavant invisibles. Cette cartographie plus précise aide les chercheurs à cibler les populations cellulaires et les voies exactes à viser pour de nouveaux médicaments ou interventions, de la formation sanguine et de l’immunité aux cellules immunitaires du cerveau dans la maladie d’Alzheimer.
Citation: Lin, Z., Su, C. scTWAS: a powerful statistical framework for single-cell transcriptome-wide association studies. Nat Commun 17, 3853 (2026). https://doi.org/10.1038/s41467-026-70374-7
Mots-clés: génomique des cellules uniques, cartographie du risque génétique, cellules immunitaires, maladie d’Alzheimer, génétique statistique