Clear Sky Science · de
scTWAS: ein leistungsfähiges statistisches Rahmenwerk für Transkriptom-weite Assoziationsstudien auf Einzelzellebene
Warum der Blick auf einzelne Zellen die Medizin verändern kann
Die meisten genetischen Studien zu Krankheiten funktionieren wie das Zuhören einer Menschenmenge: Man hört ein allgemeines Dröhnen, verpasst aber, was jede einzelne Person sagt. Dieser Artikel zeigt, wie man die Ohren auf einzelne Stimmen einstellt. Die Autorinnen und Autoren stellen scTWAS vor, eine neue Methode, die DNA-Unterschiede mit Krankheiten verbindet, indem sie die Genaktivität in bestimmten Zelltypen und noch feineren Zelluntertypen mithilfe von Einzelzell-RNA-Sequenzierungsdaten betrachtet. Dieser schärfere Blick offenbart, welche konkreten Zellen — und welche Gene in ihnen — Erkrankungen wie Blutstörungen, Autoimmunerkrankungen und Alzheimer tatsächlich antreiben.
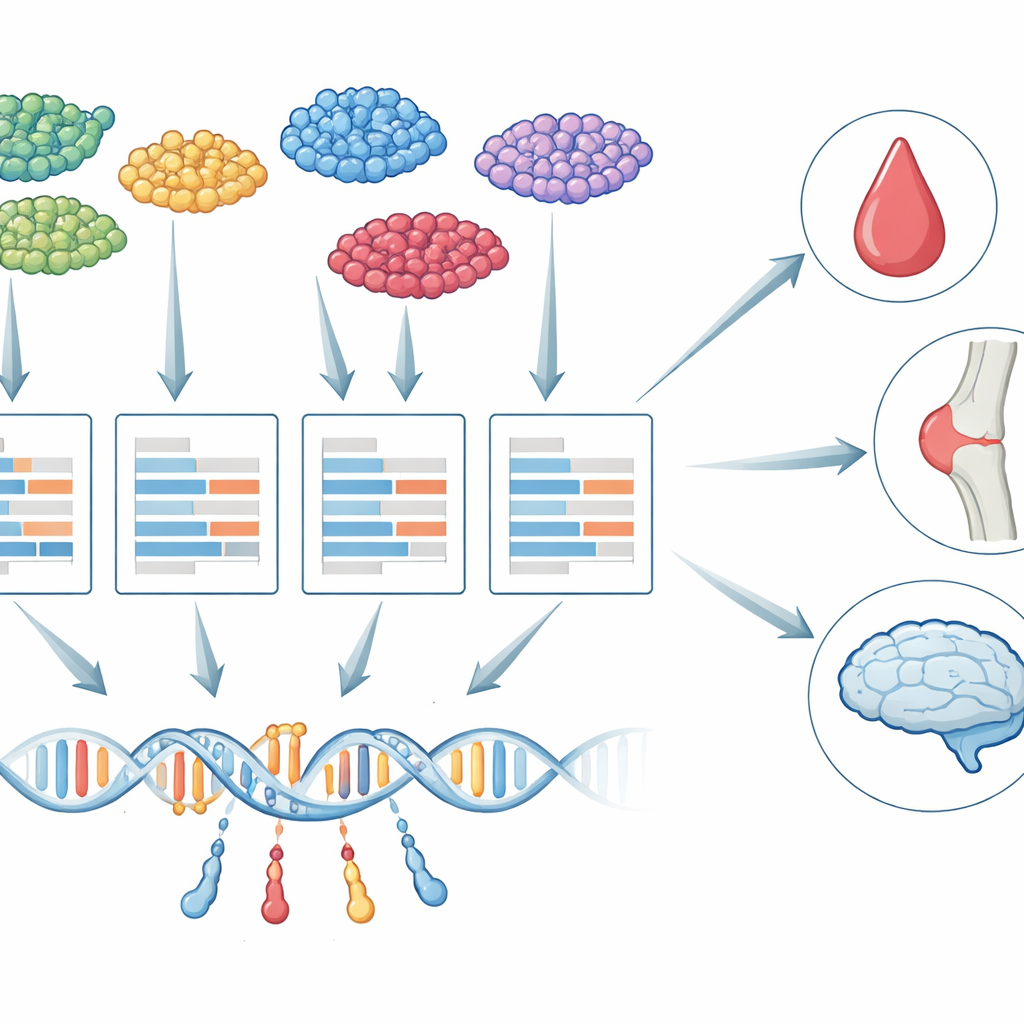
Von Gewebeproben zu Einzelzellen
Mehr als ein Jahrzehnt lang nutzten Forschende eine Strategie namens Transkriptom-weite Assoziationsstudien (TWAS), um genetische Varianten mit Krankheiten zu verknüpfen. TWAS arbeitet in zwei Schritten: Zuerst lernt man, wie DNA-Veränderungen die Genaktivität beeinflussen, dann prüft man, ob die genetisch vorhergesagte Aktivität jedes Gens mit einem Merkmal wie der Thrombozytenzahl oder dem Demenzrisiko zusammenhängt. Bislang beruhte fast alle TWAS-Forschung auf „Bulk“-Gewebeproben, bei denen RNA aus vielen Zelltypen vermischt ist. Diese Vermischung verschleiert wichtige Unterschiede: die genetische Steuerung eines Gens in einer Mikroglia-Zelle im Gehirn kann sich stark von derselben Genregulation in einem Neuron unterscheiden, und nur einige dieser Zellen können für eine bestimmte Krankheit tatsächlich relevant sein.
Das Problem rauschhafter Einzelzelldaten
Neue, populationsweite Einzelzell-RNA-Sequenzierungen machen es jetzt möglich, tausende Einzelzellen pro Person und über viele Personen hinweg zu messen. Diese Daten sind jedoch unordentlich: Zählwerte sind dünn verteilt (viele Nullen), werden stark von technischen Besonderheiten des Experiments beeinflusst und variieren stark von Zelle zu Zelle, selbst wenn die biologische Grundlage gleich ist. Frühere Versuche, Einzelzelldaten in TWAS einzubringen, verwendeten ad-hoc-Normalisierungstricks aus der Bulk-RNA-Methodik in der Hoffnung, das Rauschen zu bändigen. Die Autorinnen und Autoren zeigen, dass solche Abkürzungen die wahren genetischen Effekte auf die Genaktivität verzerren können, was zu schwächeren Prognosen und weniger entdeckten Gen–Krankheits-Verknüpfungen führt — besonders in seltenen oder schwierigen Zelltypen.
Wie scTWAS das Signal reinigt
scTWAS begegnet diesen Herausforderungen, indem es Biologie und Messfehler explizit trennt. Zunächst aggregiert die Methode Einzelzell-Zählungen pro Person und Zelltyp zu einem „Pseudo-Bulk“-Profil, wodurch die Sparse reduziert wird, ohne die Zelltyp-Identität zu verlieren. Anschließend nutzt sie ein zweischichtiges statistisches Modell: Eine Schicht beschreibt, wie DNA-Varianten und grundlegende Merkmale wie Alter die wahre zugrunde liegende Genaktivität einer Person in einem bestimmten Zelltyp beeinflussen; die andere Schicht modelliert, wie das Sequenziergerät diese Aktivität in verrauschte Zählwerte übersetzt, einschließlich Effekten variierender Sequenziertiefen. Durch das Anpassen dieses Modells mit einem spezialisierten gewichteten Regressionsalgorithmus gewichtet scTWAS die rauschhaftesten Proben ab und schätzt die genetisch regulierte Expression für jedes Gen in jedem Zelltyp genauer.
Krankheitsgene dort finden, wo sie wirklich wirken
Sobald diese zelltyp-spezifischen Vorhersagemodelle trainiert sind, setzt scTWAS sie in großen genomweiten Assoziationsstudien ein, um auf Gen–Merkmals-Verknüpfungen zu testen. In simulierten Daten, die echte Einzelzell-Experimente nachahmen, übertrifft scTWAS bestehende Methoden durchweg in Vorhersagegenauigkeit und Teststärke, mit besonders großen Vorteilen für seltene Zelltypen, in denen Daten knapp sind. Bei Anwendung des Rahmens auf Immunzellen zeigen die Autorinnen und Autoren, dass scTWAS deutlich mehr Gene identifiziert, die mit 29 Blutmerkmalen sowie mit rheumatoider Arthritis, Lupus und Asthma assoziiert sind. Viele dieser Signale heben bestimmte Immunzelltypen hervor — etwa spezifische Monozyten- oder T-Zell-Untergruppen — als Hauptbühne, auf der bestimmte Gene das Krankheitsrisiko beeinflussen; einige Assoziationen wurden in Bulk-Blut-Analysen vollständig übersehen.
Blick in Gehirn-Zelluntertypen bei Alzheimer
scTWAS erweist sich im Gehirn als noch aufschlussreicher. Mit Einzelkern-Daten aus Hunderten gespendeter menschlicher Gehirne bauten die Autorinnen und Autoren Vorhersagemodelle für sechs wichtige Gehirnzelltypen und 75 feinere Untertypen. Diese kombinierten sie dann mit genetischen Daten zu Alzheimer, um auf zellulärer Auflösung zu kartieren, wo Risikogene wahrscheinlich wirken. Manche Gene treten in vielen Zelltypen auf, was auf breit gefächerte Rollen im Gehirn hindeutet, während andere auffällig spezifisch sind. Beispielsweise zeigt ein bekanntes Risikogen, MS4A6A, eine starke Assoziation nur in einem krankheitsassoziierten Mikroglia-Untertyp, der mit Lipidverarbeitung verbunden ist, und PPP1R37 ist nur in einem inflammatorischen Mikroglia-Untertyp in der Nähe der bekannten APOE-Risikoregion assoziiert. Diese Muster deuten auf unterschiedliche Mikroglia-Zustände als Schlüsselakteure darin hin, wie bestimmte genetische Varianten das Alzheimer-Risiko vorantreiben.

Was das für künftige Therapien bedeutet
Für Nicht-Spezialisten ist die Kernbotschaft, dass der Ort, an dem ein Gen wirkt, ebenso wichtig sein kann wie die Funktion des Gens. Indem TWAS von vermischten Geweben auf präzise Zelltypen und Untertypen verlagert wird und indem die Besonderheiten von Einzelzell-Messungen sorgfältig modelliert werden, bringt scTWAS Gen–Krankheits-Verbindungen ans Licht, die zuvor unsichtbar waren. Diese schärfere Karte hilft Forschenden, genau die Zellpopulationen und Signalwege zu identifizieren, die für neue Medikamente oder Interventionen ins Visier genommen werden sollten — von Blutbildung und Immunität bis zu den Immunzellen des Gehirns bei Alzheimer-Krankheit.
Zitation: Lin, Z., Su, C. scTWAS: a powerful statistical framework for single-cell transcriptome-wide association studies. Nat Commun 17, 3853 (2026). https://doi.org/10.1038/s41467-026-70374-7
Schlüsselwörter: Einzelzell-Genomik, genetische Risiko-Kartierung, Immunzellen, Alzheimer-Krankheit, statistische Genetik