Clear Sky Science · sv
IFI16 är avgörande för att koppla DNA-skada till ferroptos vid akut njurskada
Varför njurstress och celldöd spelar roll
Akut njurskada är en plötslig förlust av njurfunktion som ofta drabbar patienter på sjukhus efter större operationer, infektioner eller lågt blodtryck. Den är vanlig, farlig och behandlas i dag mest med stödjande åtgärder som vätska och dialys snarare än riktade läkemedel. Denna studie avslöjar en tidigare dold kedja av händelser inne i njurtubuliceller som kopplar skada på deras DNA till en nyare form av järndriven celldöd kallad ferroptos. Genom att peka ut en enskild “strömbrytarmolekyl” föreslår arbetet nya sätt att skydda njurar när blodflödet avbryts och sedan återställs, som sker vid många medicinska nödsituationer.
En dold trigger inne i njurceller
Forskarna fokuserade på cellerna som bekläder njurens små tuber, vilka är särskilt sårbara när blod- och syretillförseln avbryts och sedan återupptas, en process som kallas ischemia/reperfusion. De riktade in sig på ett protein kallat IFI16 (och dess musmotsvarighet p204), som normalt hjälper celler att känna igen främmande eller skadat DNA och avgöra om de ska reparera, initiera inflammation eller självförstöra. Vid undersökning av njurbiopsier från patienter med akut tubulär nekros, en svår form av akut njurskada, fann de mycket högre nivåer av IFI16 i tubuliceller jämfört med friska njurar, och mängden IFI16 korrelerade med blodmarkörer för nedsatt njurfunktion. Hos möss som utsattes för njurishemi/reperfussion steg den muslika p204 över tid, särskilt i cellkärnorna i proximala tubuliceller, den del som tar störst skada. 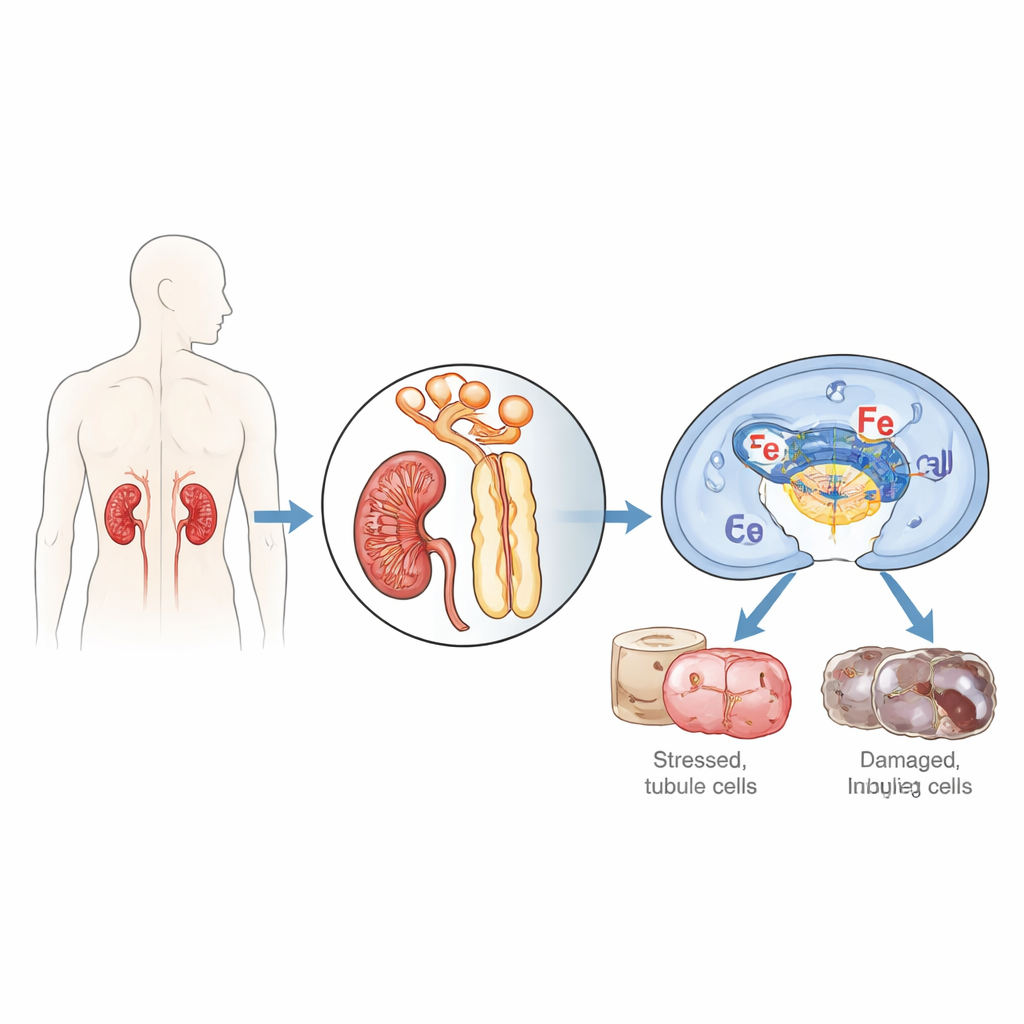
Att stänga av p204 lindrar njurskada
För att testa om detta protein aktivt skadar njuren eller bara är en förbipasserande markör konstruerade gruppen möss vars tubuliceller specifikt saknade p204. Dessa djur var i övrigt friska, men när deras njurar utsattes för ischemia/reperfusion klarade de sig mycket bättre än normala möss. Blodprover visade lägre kreatinin och urea, vilket indikerade bättre njurfiltrering. Mikroskopiskt hade deras tubuli mindre strukturell skada och färre stressmarkörer, och infiltrationen av inflammatoriska immunceller var minskad. I odlade mänskliga njurtubuliceller gjorde borttagning av IFI16 med CRISPR-genterapi också cellerna mer motståndskraftiga mot skada från låg syrehalt följt av reoxygenering, vilket minskade både oreglerad, nekrotisk celldöd och programmerad apoptos.
Järn, fetter och en destruktiv form av celldöd
Utöver klassisk apoptos visade författarna att IFI16 och p204 kraftigt främjar ferroptos, en form av celldöd driven av järn och oxidativ skada på fetter i cellmembran. I skadade musnjurar minskade förlust av p204 kemiska tecken på lipid-skada, såsom 4-hydroxynonenal och malondialdehyd, och dämpade nivåer av ACSL4, ett enzym som laddar sårbara fettsyror in i membranen. I mänskliga tubuliceller minskade borttagning av IFI16 uppbyggnaden av lipidperoxider, ökade skyddande antioxidantssystem (inklusive glutationvägen och enzymet GPX4) och återställde balansen mellan reducerat och oxiderat glutation. Det begränsade också ökningen av fritt järn inne i cellerna genom att bevara järnlagrande proteiner och hjälpa en metallsensor att förflytta sig in i kärnan. När forskarna använde läkemedel för att blockera olika dödsvägar hade hämning av ferroptos starkast effekt i att rädda celler med överuttryckt IFI16, vilket framhäver ferroptos som huvudvägen för förstörelse som detta protein driver.
En molekylär kedja från DNA-skada till ferroptos
Genom att gå djupare kartlade studien hur IFI16 fungerar som en reläkoppling mellan skadat DNA och ferroptos. Efter stress liknande ischemia/reperfusion binder IFI16 till PARP-1, en viktig förstarespondent på DNA-brott, och förstärker dess aktivitet. Detta hyperaktiverar en DNA-skaderespons centrerad kring kinaset ATM och den välkända vakthunden p53. Genom denna axel förstärker IFI16 signaler som tömmer cellernas energibärare, ökar järntillgänglighet, försvagar antioxidantförsvar och gynnar oxidativ membranskada. Kemiska hämmare av PARP-1 eller ATM bröt denna skadliga kedja: de återställde antioxidant- och järnbuffrande proteiner, minskade järn- och lipidperoxidation och minskade celldöd, även när IFI16-nivåerna var höga. Strukturella experiment visade vidare att både DNA-bindande HIN-domänerna och den proteininteragerande PYRIN-domänen i IFI16 krävs för att engagera PARP-1, förstärka DNA-skaderesponsen och driva ferroptos. 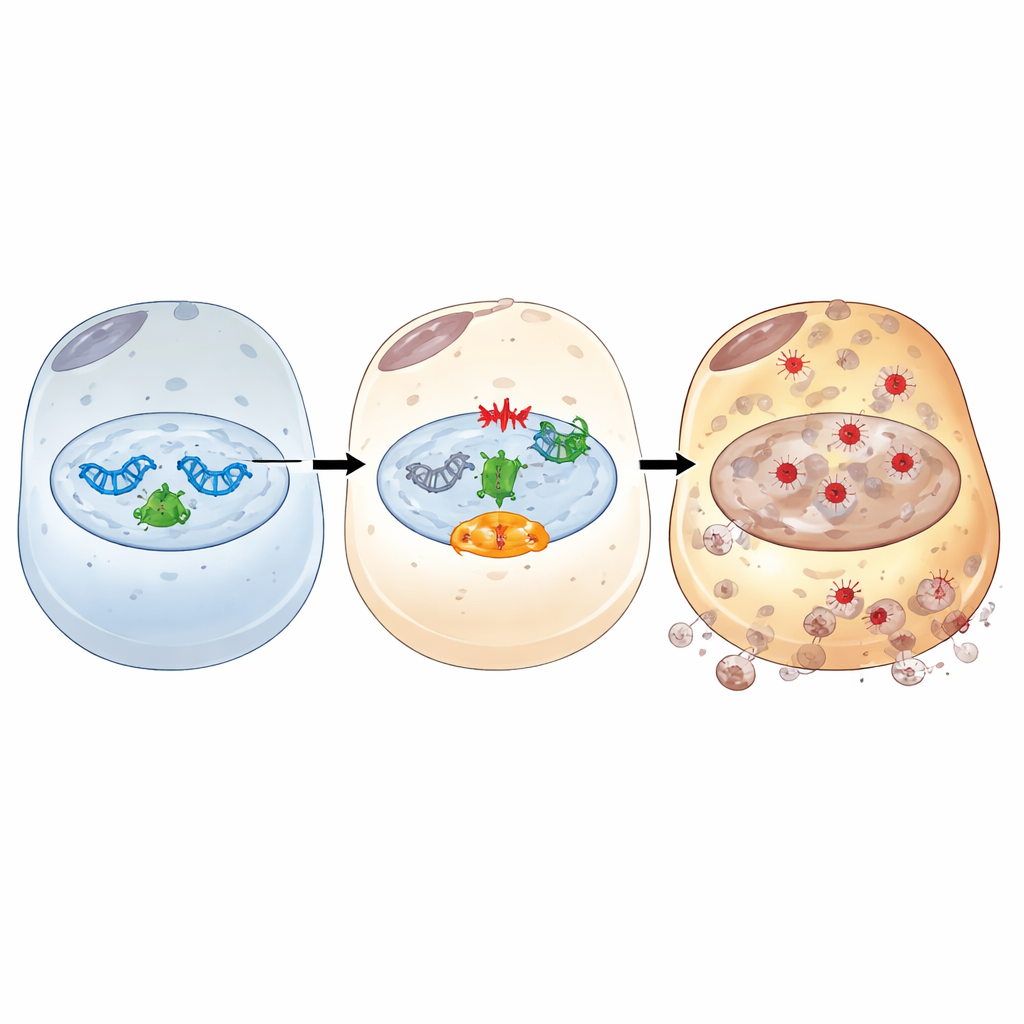
Nytt hopp för att skydda skadade njurar
Tillsammans placerar dessa fynd IFI16/p204 som en central nav som förvandlar DNA-skada i njurtubuliceller till en våg av järndriven förstörelse. Istället för att brett stänga ner DNA-reparationsmaskineriet — vilket kan förvärra njurutfallet — föreslår studien en mer precis strategi: att dämpa den överdrivna, IFI16-drivna armen av skaderesponsen som tippar celler från överlevnad mot död. I praktiska termer skulle behandlingar som minskar IFI16-aktivitet, blockerar dess interaktion med PARP-1 eller stör dess förmåga att binda skadat DNA kunna mildra konsekvenserna av akut njurskada, begränsa ferroptos samt andra sammanflätade dödsvägar. Även om sådana behandlingar återstår att utveckla och pröva på människor, ritar detta arbete en tydlig molekylär färdplan mot riktade njurskyddande läkemedel.
Citering: Qiao, Z., Zhou, D., Zhang, T. et al. IFI16 is essential to linking DNA damage and ferroptosis in acute kidney injury. Cell Death Dis 17, 350 (2026). https://doi.org/10.1038/s41419-026-08604-5
Nyckelord: akut njurskada, ferroptos, DNA-skaderespons, njurtubuliceller, IFI16