Clear Sky Science · en
IFI16 is essential to linking DNA damage and ferroptosis in acute kidney injury
Why kidney stress and cell death matter
Acute kidney injury is a sudden loss of kidney function that often strikes people in the hospital after major surgery, infections, or low blood pressure. It is common, dangerous, and currently treated mostly with supportive measures like fluids and dialysis rather than true targeted drugs. This study uncovers a previously hidden chain of events inside kidney tubule cells that links damage to their DNA with a newer form of iron-driven cell death called ferroptosis. By pinpointing a single “switch” molecule, the work suggests new ways to protect kidneys when blood flow is cut off and then restored, as happens during many medical emergencies.
A hidden trigger inside kidney cells
The researchers focused on cells that line the kidney’s tiny tubes, which are especially vulnerable when blood and oxygen supply are interrupted and then restarted, a process known as ischemia/reperfusion. They homed in on a protein called IFI16 (and its mouse counterpart p204), which normally helps cells sense foreign or damaged DNA and decide whether to repair, inflame, or self-destruct. Examining kidney biopsies from patients with acute tubular necrosis, a severe form of acute kidney injury, they found much higher levels of IFI16 in tubule cells compared with healthy kidneys, and the amount of IFI16 tracked with blood markers of poor kidney function. In mice subjected to kidney ischemia/reperfusion, the mouse version p204 shot up over time, especially in the nuclei of proximal tubule cells, the segment that bears the brunt of injury. 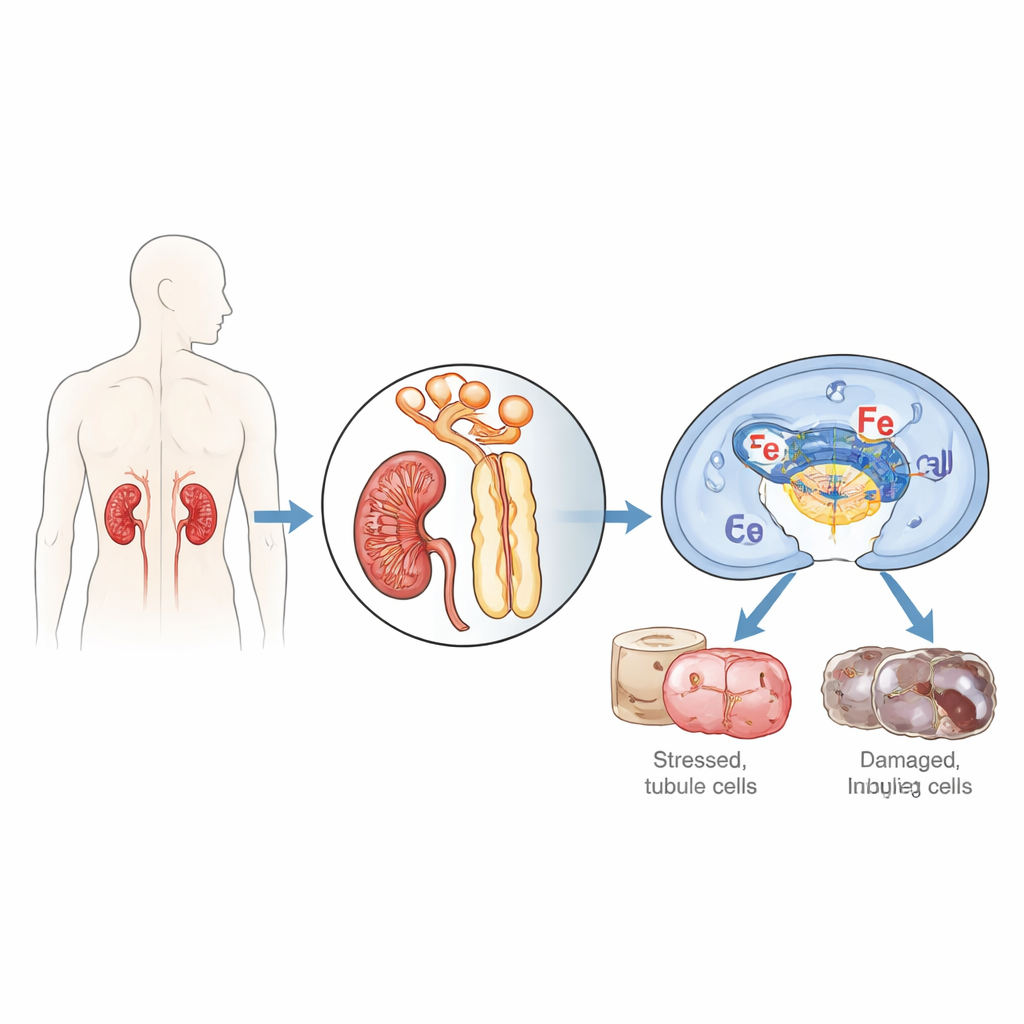
Switching off p204 eases kidney damage
To test whether this protein actively harms the kidney or is just a bystander, the team engineered mice whose tubule cells specifically lacked p204. These animals were otherwise healthy, but when their kidneys were exposed to ischemia/reperfusion, they fared far better than normal mice. Blood tests showed lower creatinine and urea, indicating better kidney filtration. Under the microscope, their tubules had less structural damage and fewer stress markers, and there was reduced invasion of inflammatory immune cells. In cultured human kidney tubule cells, deleting IFI16 using CRISPR gene editing also made the cells more resistant to injury from low oxygen followed by reoxygenation, cutting down both leaky, necrotic cell death and programmed apoptosis.
Iron, fats, and a destructive form of cell death
Beyond classic apoptosis, the authors showed that IFI16 and p204 strongly promote ferroptosis, a form of cell death fueled by iron and the oxidative damage of fats in cell membranes. In injured mouse kidneys, loss of p204 reduced chemical footprints of lipid damage, such as 4-hydroxynonenal and malondialdehyde, and dampened levels of ACSL4, an enzyme that loads vulnerable fatty acids into membranes. In human tubule cells, knocking out IFI16 lowered lipid peroxide build-up, boosted protective antioxidant systems (including the glutathione pathway and the enzyme GPX4), and restored the balance of reduced and oxidized glutathione. It also limited the surge of free iron inside cells by preserving iron-storing proteins and helping a metal-sensing factor move into the nucleus. When the researchers used drugs to block different death pathways, inhibiting ferroptosis had the strongest effect in rescuing IFI16-overexpressing cells, highlighting ferroptosis as the main route of destruction driven by this protein.
A molecular chain from DNA damage to ferroptosis
Diving deeper, the study mapped out how IFI16 acts as a relay between damaged DNA and ferroptosis. After ischemia/reperfusion-like stress, IFI16 binds to PARP-1, a key first responder to DNA breaks, and boosts its activity. This hyperactivates a DNA damage signaling route centered on the ATM kinase and the famous guardian protein p53. Through this axis, IFI16 amplifies signals that deplete cellular energy carriers, increase iron availability, weaken antioxidant defenses, and favor oxidative membrane damage. Chemical blockers of PARP-1 or ATM broke this harmful chain: they restored antioxidant and iron-buffering proteins, reduced iron and lipid peroxidation, and lessened cell death, even when IFI16 levels were high. Structural experiments further revealed that both the DNA-binding HIN domains and the protein-interacting PYRIN domain of IFI16 are required to engage PARP-1, enhance the DNA damage response, and drive ferroptosis. 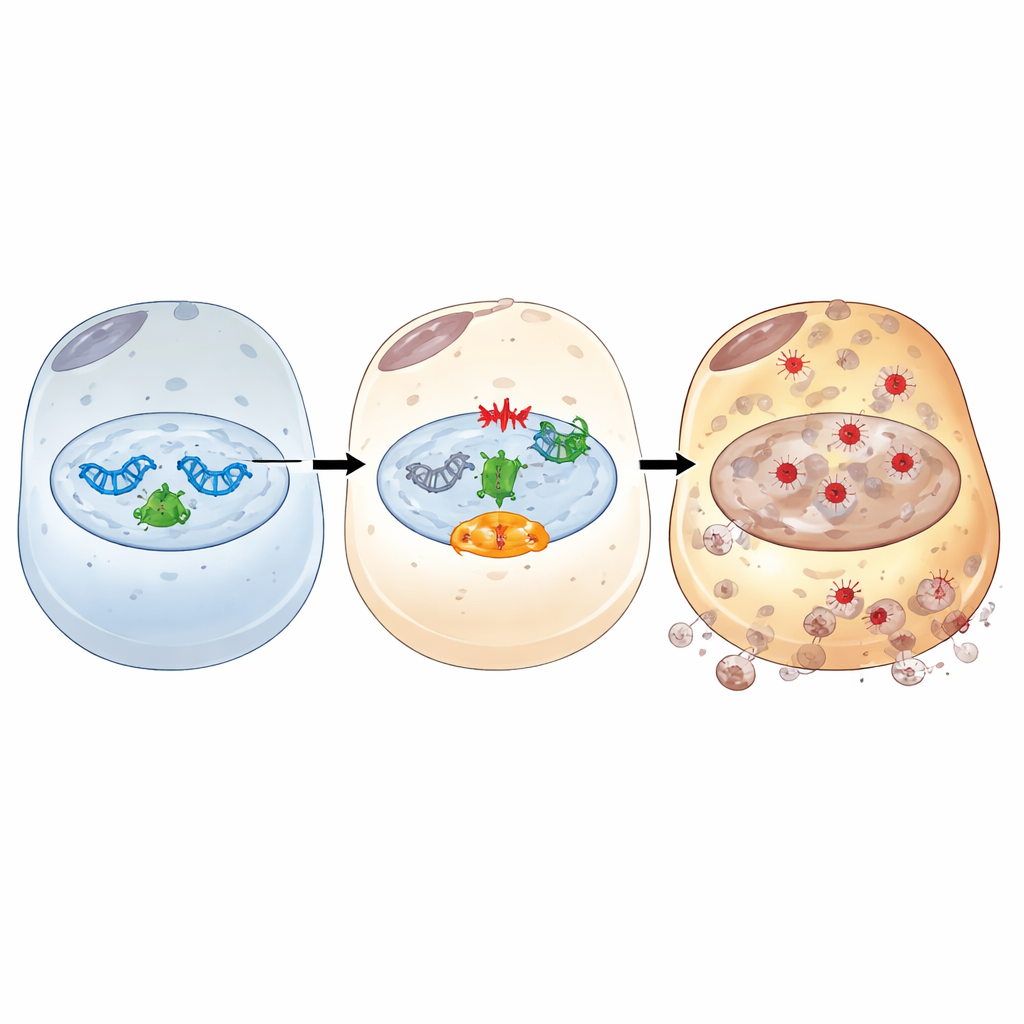
New hope for protecting injured kidneys
Together, these findings position IFI16/p204 as a central hub that turns DNA injury in kidney tubule cells into a wave of iron-fueled destruction. Rather than broadly shutting down the DNA repair machinery—which can worsen kidney outcomes—the study suggests a more precise strategy: tamping down the excessive, IFI16-driven arm of the damage response that tips cells from survival toward death. In practical terms, therapies that reduce IFI16 activity, block its interaction with PARP-1, or disrupt its ability to bind damaged DNA could soften the blow of acute kidney injury, limiting ferroptosis as well as other intertwined death pathways. While such treatments remain to be developed and tested in humans, this work charts a clear molecular roadmap toward targeted kidney-protective drugs.
Citation: Qiao, Z., Zhou, D., Zhang, T. et al. IFI16 is essential to linking DNA damage and ferroptosis in acute kidney injury. Cell Death Dis 17, 350 (2026). https://doi.org/10.1038/s41419-026-08604-5
Keywords: acute kidney injury, ferroptosis, DNA damage response, kidney tubule cells, IFI16