Clear Sky Science · sv
Dynamisk epigenetisk reglering av BCLAF1-splitsning vid akut myeloisk leukemi
Varför detta är viktigt för blodcancer
Akut myeloisk leukemi (AML) är en aggressiv blodcancer där omogna vita blodkroppar växer okontrollerat och motstår många behandlingar. Denna studie kartlägger hur en enda gen, BCLAF1, klipps och sammanfogas till olika varianter i leukemiceller — och hur läkemedel som påverkar cellens ”epigenetiska” brytare kan vända detta splitsningsmönster mot ett mer normalt, mindre aggressivt tillstånd. Att förstå detta dolda kontrollskikt kan öppna nya vägar för att tygla resistenta leukemier utan att behöva helt nya cytostatika.
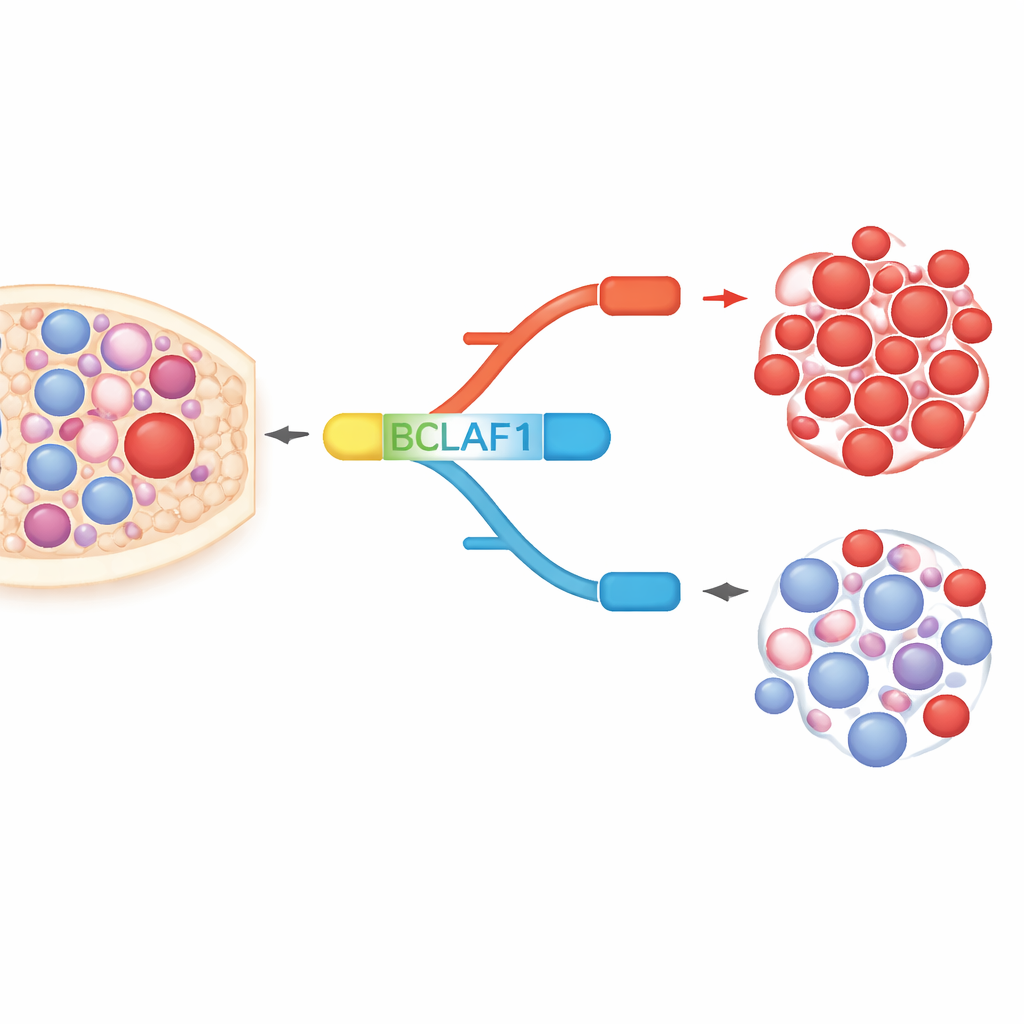
En gen, två motsatta versioner
BCLAF1 är ett multitaskande protein som är involverat i celldöd, DNA-reparation och stressrespons. Genen kan splitsas till en fullängdsform och en kortare form, som uppträder nästan som molekylära motsatser. I AML-celler och patientprover fann författarna en starkt övervikt för fullängdsvarianten, som stödjer cancercelltillväxt, medan den kortare varianten, som verkar mer skyddande, är relativt underrepresenterad. I friska blodstam- och progenitorceller hålls däremot de två formerna i balans. Denna leukemispecifika snedfördelning tyder på att fullängds-BCLAF1 både kan fungera som en sjukdomsmarkör och som en drivkraft för malign beteende.
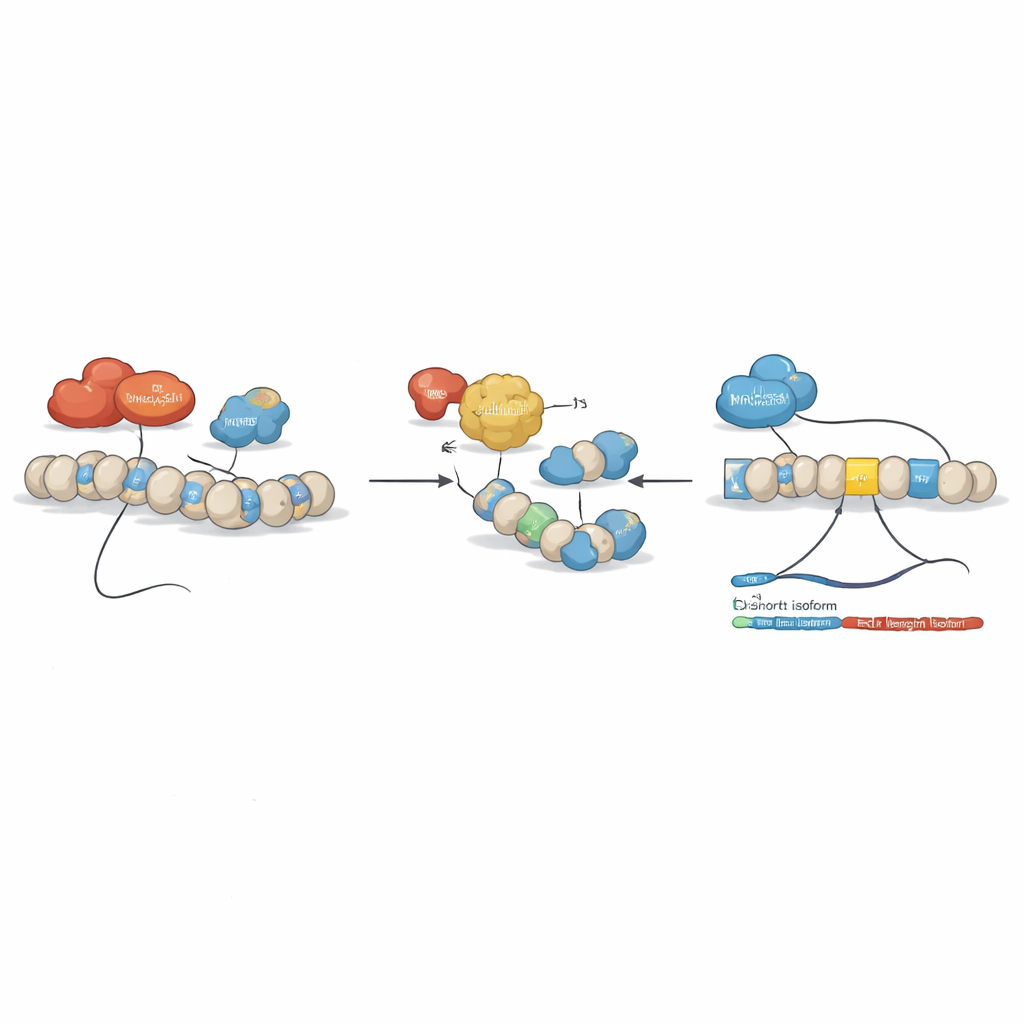
Hur kemiska brytare omformar splitsningen
Teamet undersökte sedan om epigenetiska läkemedel — föreningar som ändrar hur tätt DNA paketeras och läses — kunde återställa denna obalans. De behandlade leukemicellinjer med flera sådana ämnen, inklusive histondeacetylashämmaren SAHA (vorinostat) och föreningar som riktar DNA-metylering. Ett tydligt mönster framträdde: vissa läkemedel minskade fullängds-BCLAF1 och ökade den kortare formen, utan att påverka andra BCLAF1-varianter. Detaljerade kromatinstudier visade att under normala leukemiförhållanden sitter två enzymer, DNMT3A och HDAC1, på nyckelregioner av BCLAF1-genen och bidrar till en repressiv konfiguration som gynnar fullängds-splitset. SAHA-behandling luckrade upp denna struktur, avlägsnade DNMT3A och HDAC1, ökade aktiverande histonmarkörer och möjliggjorde att en annan faktor, MRG15, binder inom exon 5, vilket förskjuter splitsningen mot den kortare varianten.
Två DNA-metyleringsenzymer, motsatta roller
För att dissekera rollerna för de närbesläktade enzymerna DNMT3A och DNMT3B konstruerade forskarna AML-celler där vardera enzymet kunde stängas av specifikt. Nedreglering av DNMT3A bromsade celltillväxten, försvagade HDAC1-bindningen på BCLAF1-genen och drev splitsningen mot den kortare, sannolikt tumörsupprimerande formen. DNMT3B uppträdde mycket annorlunda: det är faktiskt reducerat i AML-patientprover, och när teamet ytterligare minskade DNMT3B i celler ökade detta oväntat den cancerfrämjande fullängds-BCLAF1-isoformen. Biokemiska tester visade att DNMT3B kan binda direkt till BCLAF1-RNA snarare än bara till DNA, och efter SAHA-behandling eller förlust av DNMT3A interagerar det också med den korta RNA-formen. Tillsammans föreslår dessa fynd att DNMT3A formar BCLAF1-splitsning huvudsakligen genom kromatinstruktur, medan DNMT3B finjusterar processen genom att interagera med själva RNA:t.
Finjustering av BCLAF1 efter att det har bildats
Berättelsen slutar inte när BCLAF1-proteinet har producerats. Författarna undersökte hur kemiska märken som läggs på BCLAF1 efter translation — särskilt fosforylering — förändras som svar på epigenetiska läkemedel och hur detta påverkar proteinets lokalisering i cellen. Vid SAHA-behandling observerade de förändringar i fosforylering på specifika platser och i om BCLAF1 satt i kärnan eller läckte ut i cytoplasman. Storskaliga proteomiska och fosfoproteomiska analyser visade att SAHA brett dämpar vägar som är involverade i celltillväxt, DNA-replikation och RNA-processning, inklusive spliceosomets maskineri som hjälper till att klippa och sammanfoga RNA. Dessa data stödjer idén att BCLAF1 utgör en knutpunkt som länkar kromatintillstånd, RNA-splitsning och cellcykelkontroll.
Vad detta innebär för framtida leukemibehandling
Sammanfattningsvis visar studien att AML-celler utnyttjar epigenetisk maskineri för att favorisera en cancerfrämjande version av BCLAF1, samtidigt som en kortare, mer skyddande form undertrycks. Genom att störa nyckelenzymer som DNMT3A, HDAC1 och DNMT3B kan epigenetiska läkemedel återbalansera denna splitsningsbeslutning och försvaga leukemicellernas överlevnadsprogram. För en icke-specialist är huvudpoängen att det inte bara spelar roll vilka gener som är på eller av, utan också hur varje gens budskap redigeras. Att rikta in sig på redigerarna — snarare än genen själv — kan erbjuda ett kraftfullt sätt att återställa hälsosammare mönster av genanvändning och förbättra utfallen för patienter med svårbehandlad AML.
Citering: Sgueglia, G., Massaro, C., Muro, A. et al. Dynamic epigenetic regulation of BCLAF1 splicing in acute myeloid leukemia. Cell Death Dis 17, 344 (2026). https://doi.org/10.1038/s41419-026-08594-4
Nyckelord: akut myeloisk leukemi, alternativ splitsning, epigenetisk terapi, BCLAF1, DNMT3A