Clear Sky Science · en
Dynamic epigenetic regulation of BCLAF1 splicing in acute myeloid leukemia
Why this matters for blood cancer
Acute myeloid leukemia (AML) is an aggressive blood cancer where immature white blood cells grow out of control and resist many treatments. This study uncovers how a single gene, BCLAF1, is cut and pasted into different versions inside leukemia cells—and how drugs that act on the cell’s “epigenetic” switches can flip this splicing pattern back toward a more normal, less aggressive state. Understanding this hidden layer of control could open new ways to tame resistant leukemias without needing entirely new chemotherapy drugs.
One gene, two opposing versions
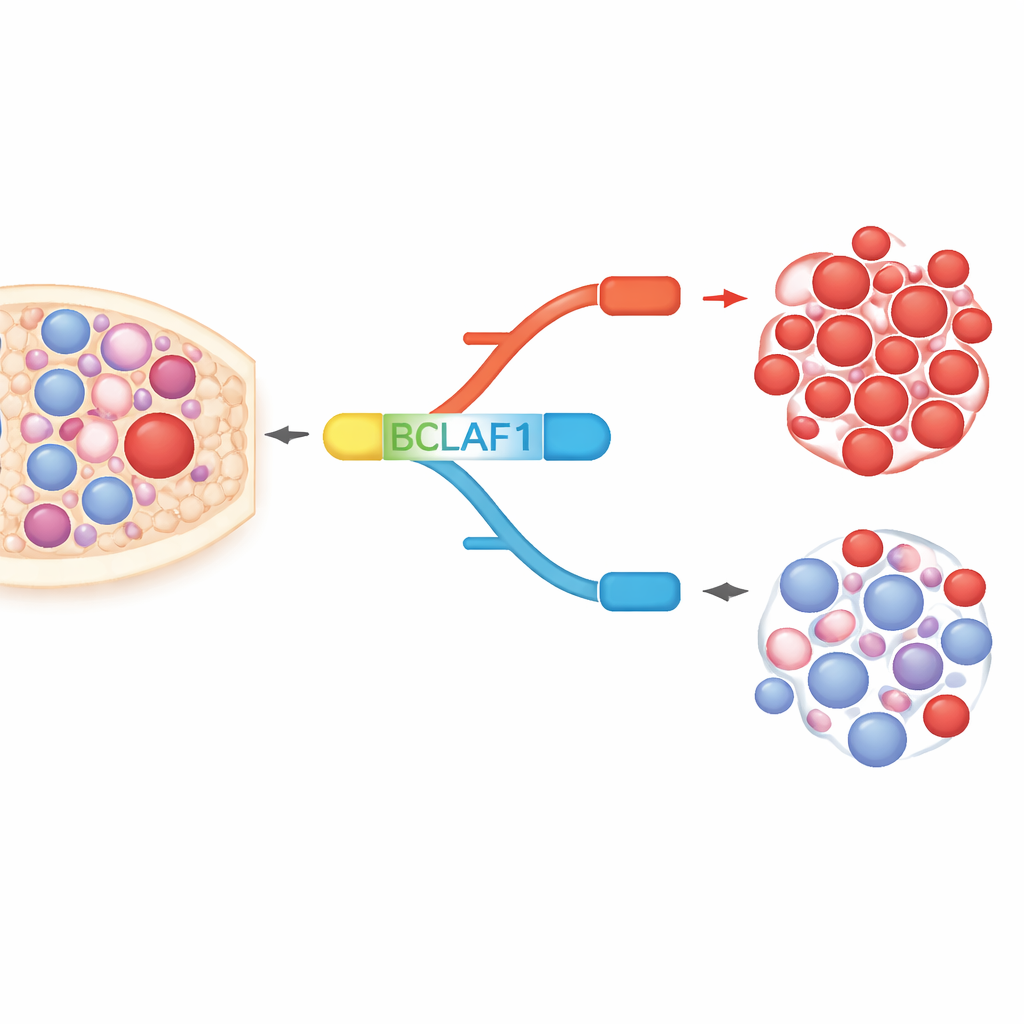
BCLAF1 is a multitasking protein involved in cell death, DNA repair, and response to stress. The gene can be spliced into a full-length form and a shorter form, which behave almost like molecular opposites. In AML cells and patient samples, the authors found a strong tilt toward the full-length version, which supports cancer cell growth, while the shorter version, which appears more protective, is relatively underrepresented. In healthy blood stem and progenitor cells, by contrast, the two forms are kept in balance. This leukemia-specific skew suggests that the full-length BCLAF1 version could serve as both a marker of disease and a driver of malignant behavior.
How chemical switches reshape splicing
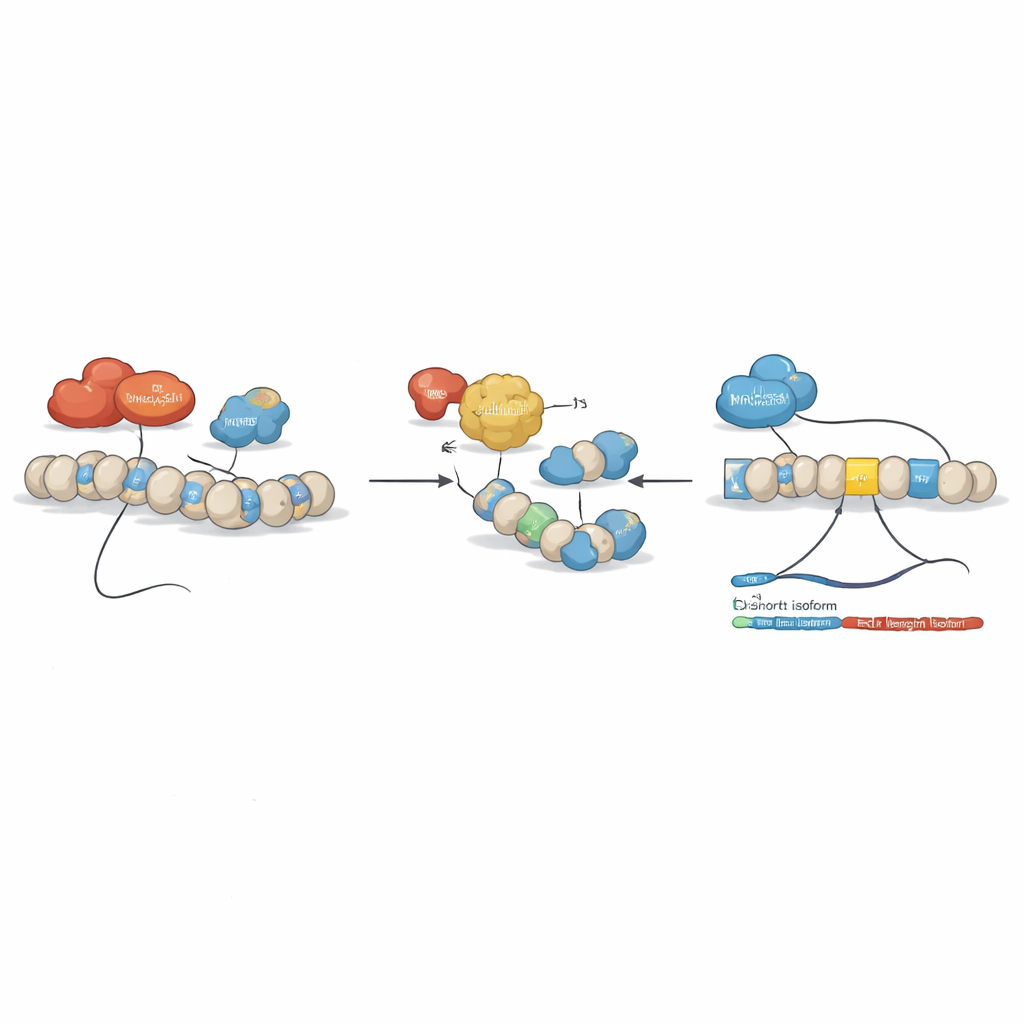
The team then asked whether epigenetic drugs—compounds that modify how tightly DNA is packaged and read—could reset this imbalance. They treated leukemia cell lines with several such agents, including the histone deacetylase inhibitor SAHA (vorinostat) and DNA methylation–targeting compounds. A clear pattern emerged: certain drugs reduced the full-length BCLAF1 form and boosted the shorter form, without affecting other BCLAF1 variants. Detailed chromatin studies showed that, under normal leukemia conditions, two enzymes, DNMT3A and HDAC1, sit on key regions of the BCLAF1 gene and help maintain a repressive configuration that favors the full-length splice. SAHA treatment loosened this structure, removed DNMT3A and HDAC1, increased activating histone marks, and allowed another factor, MRG15, to bind within exon 5, shifting splicing toward the shorter version.
Two DNA methylation enzymes, opposite roles
To dissect the roles of related enzymes DNMT3A and DNMT3B, the researchers engineered AML cells in which each enzyme could be specifically switched off. Knocking down DNMT3A slowed cell growth, weakened HDAC1 binding on the BCLAF1 gene, and drove splicing toward the shorter, likely tumor-suppressive version. DNMT3B behaved very differently: it is actually reduced in AML patient samples, and when the team further decreased DNMT3B in cells, this unexpectedly increased the cancer-promoting full-length BCLAF1 isoform. Biochemical assays revealed that DNMT3B can bind directly to BCLAF1 RNA rather than just to DNA, and after SAHA treatment or DNMT3A loss it also engages with the short RNA form. Together, these findings suggest DNMT3A shapes BCLAF1 splicing mainly through chromatin structure, while DNMT3B fine-tunes the process by interacting with the RNA itself.
Fine-tuning BCLAF1 after it is made
The story does not end once BCLAF1 protein is produced. The authors investigated how chemical tags added to BCLAF1 after translation—especially phosphorylation—change in response to epigenetic drugs and how this affects the protein’s location in the cell. With SAHA treatment, they observed shifts in phosphorylation at specific sites and changes in whether BCLAF1 sat in the nucleus or leaked into the cytoplasm. Large-scale proteomic and phosphoproteomic analyses showed that SAHA broadly dampens pathways involved in cell growth, DNA replication, and RNA processing, including the spliceosome machinery that helps cut and join RNAs. These data support the idea that BCLAF1 sits at a crossroads linking chromatin state, RNA splicing, and cell-cycle control.
What this means for future leukemia therapy
Overall, the study reveals that AML cells exploit epigenetic machinery to favor a cancer-promoting version of BCLAF1, while suppressing a shorter, more protective form. By interfering with key enzymes such as DNMT3A, HDAC1, and DNMT3B, epigenetic drugs can rebalance this splicing decision and weaken leukemia cell survival programs. For a lay observer, the key takeaway is that not only which genes are on or off matters, but also how each gene’s message is edited. Targeting the editors—rather than the gene itself—may offer a powerful way to restore healthier patterns of gene use and improve outcomes for patients with difficult-to-treat AML.
Citation: Sgueglia, G., Massaro, C., Muro, A. et al. Dynamic epigenetic regulation of BCLAF1 splicing in acute myeloid leukemia. Cell Death Dis 17, 344 (2026). https://doi.org/10.1038/s41419-026-08594-4
Keywords: acute myeloid leukemia, alternative splicing, epigenetic therapy, BCLAF1, DNMT3A