Clear Sky Science · ru
Высококачественная сборка метагеномов из нанопорных ридов с nanoMDBG
Чтение скрытой жизни в щепотке почвы
Каждая щепотка почвы или капля содержимого кишечника полна тысяч микробных видов, большинство из которых нельзя вырастить в лаборатории. Чтобы понять, кто они и чем занимаются, учёные читают их ДНК прямо из окружающей среды — это направление называется метагеномикой. В этой статье представлена nanoMDBG, новая вычислительная методика, превращающая сырые сигналы портативного секвенатора ДНК в высококачественные черновые геномы, что открывает путь к более быстрому, дешёвому и детальному картированию сложных микробных сообществ.
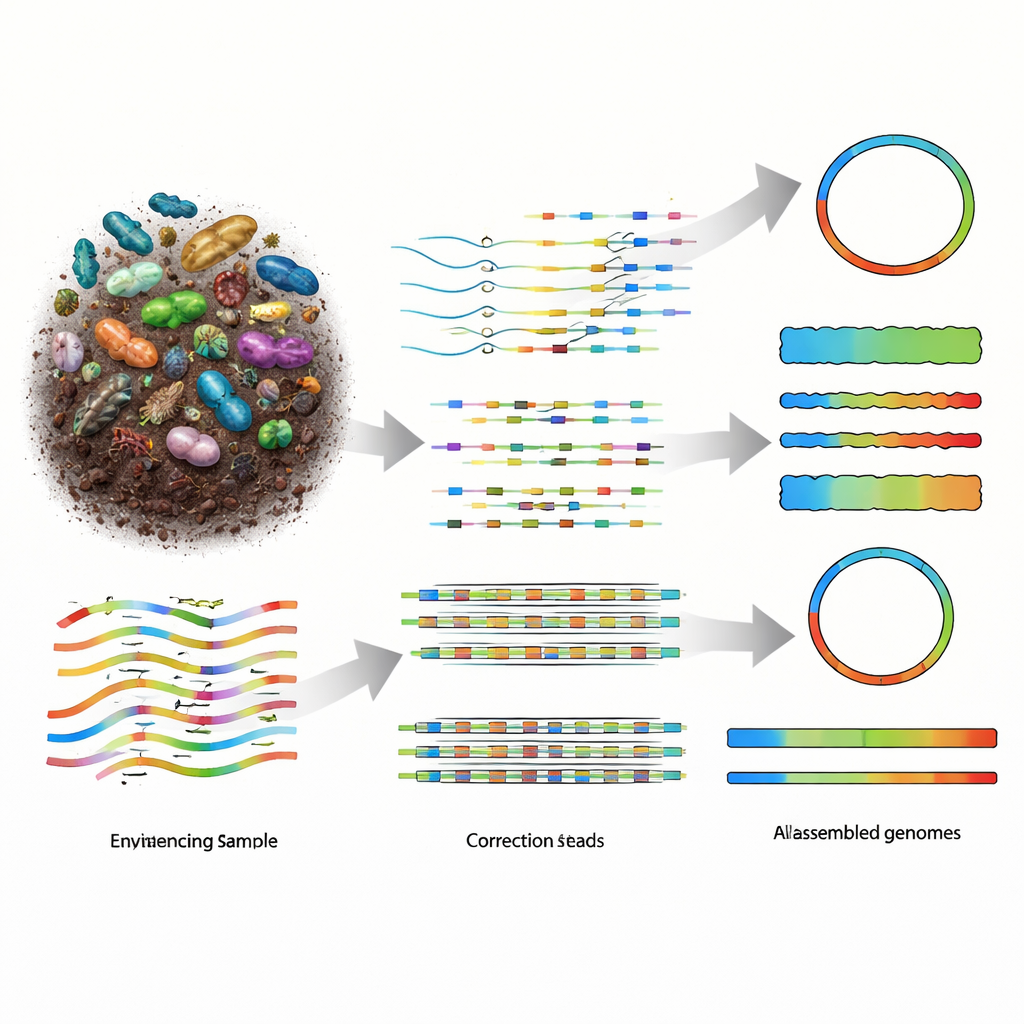
Почему восстановление геномов из природы так трудно
Метагеномика работает так: всю ДНК в образце измельчают на фрагменты, эти фрагменты секвенируют, а затем программное обеспечение собирает их обратно в геномы организмов, присутствовавших в пробе. Более старые технологии коротких ридов давали множество маленьких кусочков, что приводило к сильно фрагментированной сборке, особенно в разнообразных сообществах, например в почве. Платформы длинного чтения, включая PacBio HiFi и Oxford Nanopore Technologies (ONT), производят гораздо более длинные фрагменты, что теоретически должно облегчать реконструкцию. Риды PacBio чрезвычайно точны, но дороже, тогда как устройства ONT более доступные и портативные, но исторически давали более шумные данные. По мере того как химия ONT улучшилась до примерно одной ошибки на сто нуклеотидов, сообщество нуждалось в сборщиках, способных полноценно использовать новое сочетание длины, точности и стоимости.
От шумных сигналов к чистым строительным блокам
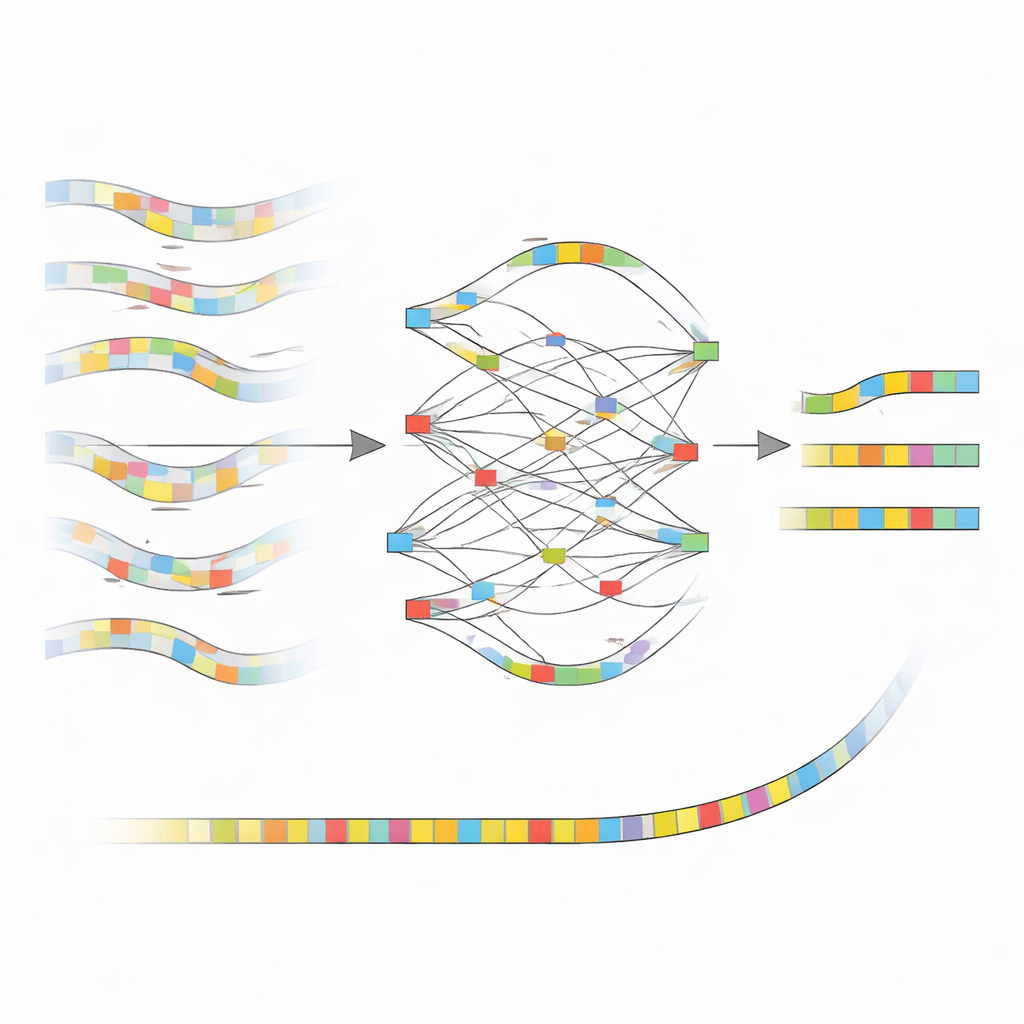
Ключевая идея nanoMDBG — работать не с каждой буквой ДНК, а с компактным наброском каждого фрагмента. Метод выбирает разреженный набор коротких ДНК-паттернов, называемых минимайзерами, из каждого рида и рассматривает упорядоченный список этих паттернов как лёгкий отпечаток. Ранее разработанное той же группой ПО metaMDBG уже использовало такие минимайзерные отпечатки для эффективной сборки очень точных ридов PacBio. Однако оставшиеся ошибки в данных ONT часто ломали эти отпечатки, вызывая разрывы и ошибочные сшивки. NanoMDBG решает эту проблему, сначала корректируя ONT-риды в уменьшенном «пространстве минимайзеров». Для каждого целевого рида алгоритм быстро набирает несколько наиболее похожих ридов, используя очень разреженные отпечатки, затем повторно проверяет их более плотными отпечатками, чтобы отсеять ложные совпадения от неродственных видов.
Как новый метод очищает картину
Когда nanoMDBG собирает надёжную группу схожих отпечатков, он наслаивает их, чтобы построить простой граф, который отслеживает, где паттерны совпадают, расходятся или показывают вставки и делеции. Вместо анализа каждой базы он оперирует только выбранными паттернами, существенно снижая вычислительную нагрузку. Наиболее поддерживаемый путь через этот граф становится консенсусным отпечатком рида, эффективно сглаживая многие исходные ошибки секвенирования. Все скорректированные отпечатки затем подаются в существующий сборщик metaMDBG, который сшивает их в более длинные фрагменты ДНК и в конце преобразует обратно в полные последовательности, после чего выполняется полировка для уточнения оставшихся мелких ошибок.
Проверка nanoMDBG на реальных микробиомах
Исследователи оценили nanoMDBG на нескольких тестовых наборах: определённая смесь из 21 известного микроорганизма, образец человеческого кишечника, эталонная смесь фекального материала человека и очень сложная аграрная почва. Они сравнили его с ведущими сборщиками длинных ридов, в частности metaFlye и прежним metaMDBG, анализируя, сколько почти полных геномов — так называемых собранных из метагенома геномов (MAG) — удалось восстановить и сколько из них получились в виде единичных непрерывных контигов. Во всех трёх реальных сообществах nanoMDBG дал значительно больше высококачественных MAG по сравнению с конкурентами и гораздо больше полных одно-контиговых геномов. В почтовом наборе данных объёмом 400 миллиардов нуклеотидов, например, он восстановил на 201 почти полный геном больше, чем metaMDBG, и на 144 больше, чем metaFlye, при этом использовал лишь часть оперативной памяти и завершил работу примерно за шесть дней вместо почти месяца.
Сравнивая дорогую точность с более дешёвыми ридами
Поскольку секвенирование ONT и PacBio проводилось на тех же образцах при сопоставимой глубине, команда могла напрямую сравнить технологии. Для образцов кишечника и стандартизированных фекальных смесей PacBio HiFi по-прежнему сохранял преимущество в общем числе геномов высшего качества, особенно при больших глубинах секвенирования. Тем не менее сборки ONT с nanoMDBG оказались удивительно близки и даже превосходили HiFi в некоторых условиях низкой глубины. Для почвенного образца, где сосуществует тысячи видов, число почти полных геномов, полученных с ONT и HiFi при большой глубине, было по сути сопоставимо, хотя HiFi чаще давал полностью непрерывные одно-контиговые геномы. Детальный анализ ошибок показал, что nanoMDBG сохранял относительно низкий уровень миссассембли и пропущенных участков покрытия и сохранял больше полноразмерных белковых генов по сравнению с конкурирующими сборщиками для ONT, особенно в сложном почвенном наборе данных.
Что это значит для изучения невидимых экосистем
Для неспециалистов главный вывод таков: дешёвые переносные секвенаторы ДНК теперь способны восстанавливать микробные геномы из сложных сред с качеством, приближающимся к более крупным и дорогим инструментам. NanoMDBG достигает этого, умно упрощая данные до повторно используемых паттернов, корректируя ошибки в этом компактном представлении и затем собирая геномы из очищенных паттернов с высокой эффективностью. Это делает возможным обследование большого числа образцов, отслеживание микробных штаммов между людьми или локациями и исследование огромного, по‑прежнему в основном неизведанного разнообразия жизни в почвах и других средах — без необходимости в ресурсах уровня суперкомпьютера. По мере дальнейшего совершенствования алгоритмов такие инструменты приблизят нас к рутинному картированию целых микробных сообществ на уровне геномов.
Цитирование: Benoit, G., James, R., Raguideau, S. et al. High-quality metagenome assembly from nanopore reads with nanoMDBG. Nat Commun 17, 3556 (2026). https://doi.org/10.1038/s41467-026-69760-y
Ключевые слова: метагеномика, нанопорное секвенирование, сборка геномов, микробиом, биоинформатика