Clear Sky Science · fr
Assemblage métagénomique de haute qualité à partir de lectures nanopore avec nanoMDBG
Lire la vie cachée dans une cuillerée de terre
Chaque pincée de sol ou goutte de liquide intestinal foisonne de milliers d'espèces microbiennes, dont la plupart ne peuvent pas être cultivées en laboratoire. Pour savoir qui elles sont et ce qu'elles font, les scientifiques lisent leur ADN directement depuis l'environnement — un domaine appelé métagénomique. Cet article présente nanoMDBG, une nouvelle méthode computationnelle qui transforme les signaux bruts d'un séquenceur d'ADN portable en génomes provisoires de haute qualité, ouvrant la voie à la cartographie de mondes microbien complexes plus rapidement, à moindre coût et avec un niveau de détail bien supérieur à celui d'avant.
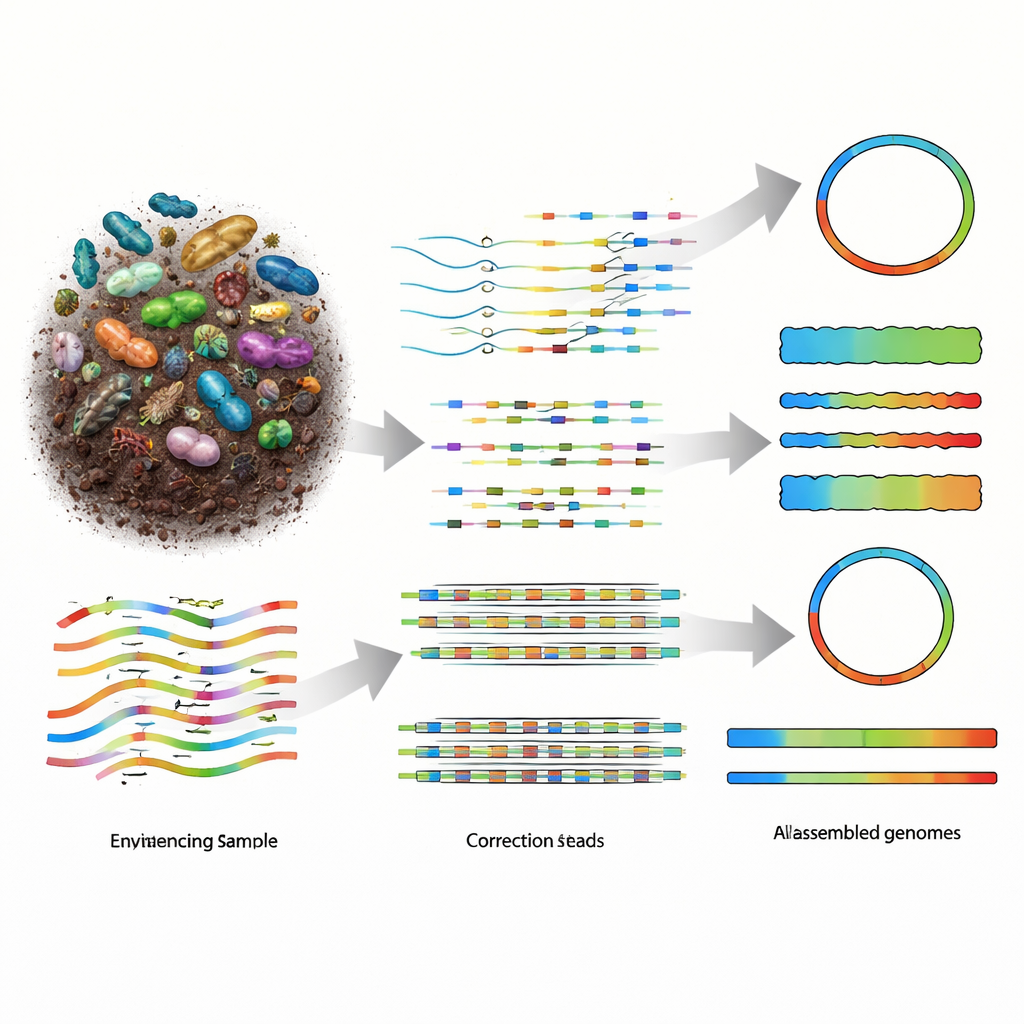
Pourquoi reconstituer des génomes à partir de la nature est si difficile
La métagénomique consiste à fragmenter tout l'ADN d'un échantillon en longs fragments, à séquencer ces fragments, puis à utiliser des logiciels pour les réassembler en génomes des organismes présents. Les technologies de lectures courtes plus anciennes fournissaient de nombreux petits morceaux, mais les puzzles résultants étaient très fragmentés, en particulier dans des communautés diversifiées comme le sol. Les plateformes de séquençage à longues lectures, notamment PacBio HiFi et Oxford Nanopore Technologies (ONT), produisent des fragments beaucoup plus longs, ce qui devrait faciliter la reconstruction. Les lectures PacBio sont extrêmement précises mais plus coûteuses, tandis que les appareils ONT sont plus abordables et très portables, mais produisaient historiquement des données plus bruitées. À mesure que la chimie ONT s'est améliorée pour atteindre environ une erreur pour cent bases, le domaine avait besoin d'assembleurs capables d'exploiter pleinement ce nouveau compromis entre longueur, précision et coût.
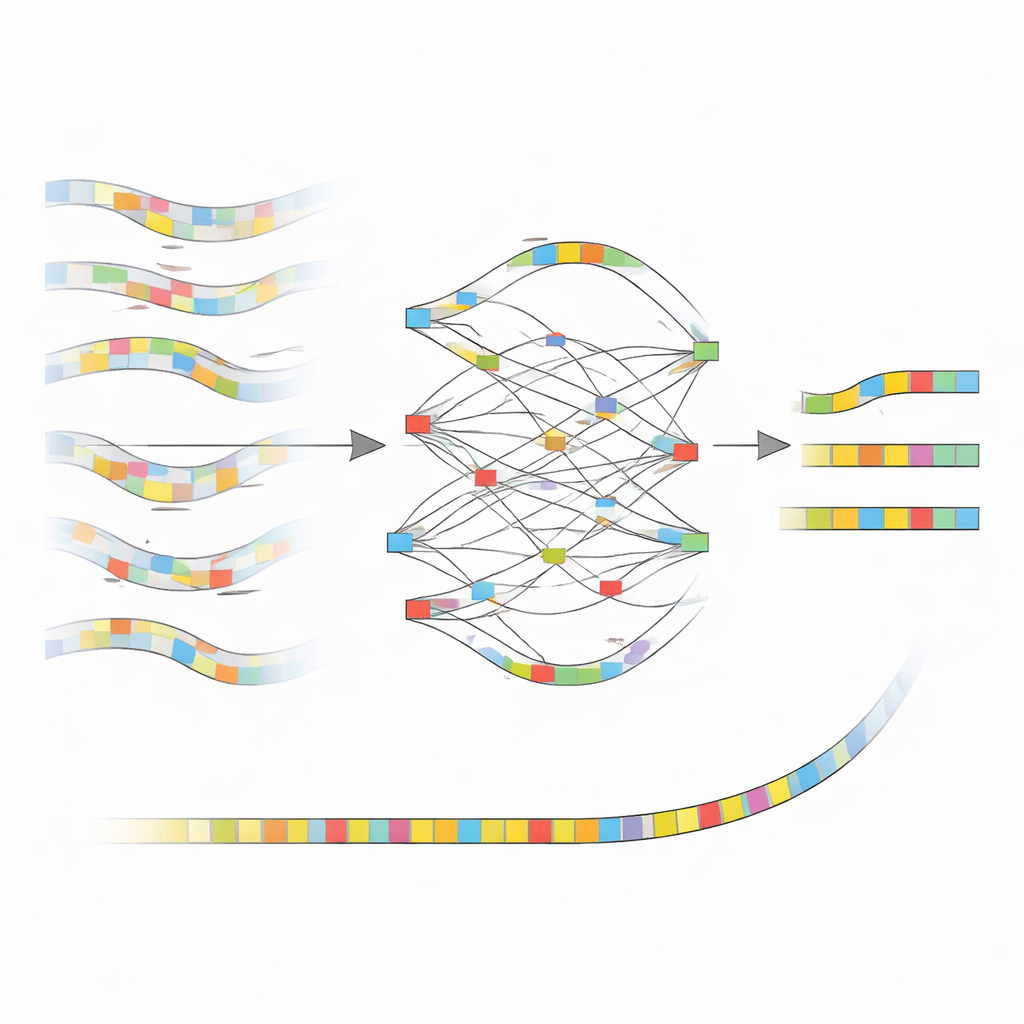
Des signaux bruités aux blocs de construction propres
L'idée centrale de nanoMDBG est de travailler avec une esquisse compacte de chaque fragment d'ADN plutôt qu'avec chaque lettre. La méthode sélectionne un ensemble parcimonieux de courts motifs d'ADN, appelés minimizers, dans chaque lecture et considère la liste ordonnée de ces motifs comme une empreinte légère. Un logiciel antérieur du même groupe, metaMDBG, utilisait déjà ces empreintes de minimizers pour assembler efficacement des lectures PacBio très précises. Cependant, les erreurs résiduelles dans les données ONT avaient tendance à casser ces empreintes, provoquant des lacunes et des jonctions erronées. NanoMDBG s'attaque à cela en corrigeant d'abord les lectures ONT dans cet « espace minimizer » réduit. Pour chaque lecture cible, l'algorithme recrute rapidement une poignée des lectures les plus similaires en utilisant des empreintes très clairsemées, puis les réexamine avec des empreintes plus denses pour éliminer les appariements fallacieux provenant d'espèces non apparentées.
Comment la nouvelle méthode clarifie le tableau
Une fois que nanoMDBG a rassemblé un groupe fiable d'empreintes similaires, il les superpose pour construire un simple graphe qui suit où les motifs concordent, divergent ou présentent des insertions et des délétions. Plutôt que d'examiner chaque base, il travaille uniquement avec ces motifs sélectionnés, réduisant fortement la charge de calcul. Le chemin le mieux soutenu à travers ce graphe devient une empreinte consensuelle pour la lecture, effaçant ainsi de nombreuses erreurs de séquençage initiales. Toutes les empreintes corrigées sont ensuite fournies à l'assembleur existant metaMDBG, qui les assemble en fragments d'ADN plus longs puis les reconvertit en séquences complètes, suivies d'une étape de polissage pour affiner les petites erreurs restantes.
Tester nanoMDBG sur des microbiomes réels
Les chercheurs ont évalué nanoMDBG sur plusieurs bancs d'essai : un mélange défini de 21 microbes connus, un échantillon de microbiote intestinal humain, un mélange de référence de matière fécale humaine, et un sol agricole très complexe. Ils ont comparé ses performances avec celles des assembleurs longue lecture de référence, notamment metaFlye et l'ancien metaMDBG, en regardant combien de génomes quasi-complets — appelés MAG (metagenome-assembled genomes) — ils pouvaient récupérer, et combien d'entre eux étaient capturés comme fragments continus uniques. Dans les trois communautés réelles, nanoMDBG a produit sensiblement plus de MAGs de haute qualité que les outils concurrents, et bien plus de génomes complets en un seul contig. Dans le jeu de données du sol de 400 milliards de bases, par exemple, il a récupéré 201 génomes quasi-complets de plus que metaMDBG et 144 de plus que metaFlye, tout en n'utilisant qu'une fraction de la mémoire et en terminant en environ six jours au lieu d'à peu près un mois.
Rivaliser avec la précision coûteuse grâce à des lectures moins chères
Parce que les séquenceurs ONT et PacBio ont été exécutés sur les mêmes échantillons à des profondeurs équivalentes, l'équipe a pu comparer directement les technologies. Pour les communautés intestinales et fécales standardisées, PacBio HiFi gardait encore un avantage sur le nombre total de génomes de très haute qualité, surtout à des profondeurs de séquençage élevées. Pourtant, les données ONT assemblées avec nanoMDBG se sont révélées étonnamment proches, et ont même dépassé HiFi dans certaines conditions de faible profondeur. Pour l'échantillon de sol, où des milliers d'espèces coexistent, les nombres de génomes quasi-complets obtenus à partir d'ONT et de HiFi étaient essentiellement comparables à haute profondeur, bien que HiFi atteignît plus souvent des génomes complètement continus en un seul contig. Des analyses d'erreurs détaillées ont montré que nanoMDBG maintenait relativement faibles les mésassemblages et les régions à couverture manquante et préservait davantage de gènes codant pour des protéines en longueur complète que les assembleurs ONT concurrents, notamment dans le difficile jeu de données provenant du sol.
Ce que cela signifie pour l'exploration des écosystèmes invisibles
Pour les non-spécialistes, le message clé est que des séquenceurs d'ADN bon marché et déployables sur le terrain peuvent désormais reconstruire des génomes microbiens d'environnements complexes à une qualité proche de celle d'instruments plus gros et plus coûteux. NanoMDBG y parvient en simplifiant intelligemment les données en motifs réutilisables, en corrigeant les erreurs dans cette représentation compacte, puis en assemblant les génomes à partir des motifs nettoyés avec une grande efficacité. Cela rend possible l'analyse d'un grand nombre d'échantillons, le suivi de souches microbiennes entre personnes ou lieux, et l'exploration de l'énorme diversité encore largement inexplorée des sols et autres habitats, le tout sans ressources de type supercalculateur. À mesure que les algorithmes continueront de s'améliorer, de tels outils nous rapprocheront de cartes de communautés microbiennes à l'échelle du génome, de manière routinière.
Citation: Benoit, G., James, R., Raguideau, S. et al. High-quality metagenome assembly from nanopore reads with nanoMDBG. Nat Commun 17, 3556 (2026). https://doi.org/10.1038/s41467-026-69760-y
Mots-clés: métagénomique, séquençage nanopore, assemblage de génomes, microbiome, bioinformatique