Clear Sky Science · de
Hochwertige Metagenom‑Assemblierung aus Nanopore‑Reads mit nanoMDBG
Verborgene Leben in einem Löffel Erde lesen
Jede Prise Erde oder jeder Tropfen Darmflüssigkeit ist voller Tausender mikrobieller Arten, von denen die meisten im Labor nicht kultivierbar sind. Um zu verstehen, was sie sind und was sie tun, lesen Forschende deren DNA direkt aus der Umwelt — ein Gebiet, das als Metagenomik bekannt ist. Dieser Artikel stellt nanoMDBG vor, eine neue rechnerische Methode, die rohe Signale eines tragbaren DNA‑Sequenzierers in hochwertige Genom‑Entwürfe verwandelt und so die Kartierung komplexer mikrobieller Welten schneller, kostengünstiger und mit deutlich größerer Detailtiefe ermöglicht als zuvor.
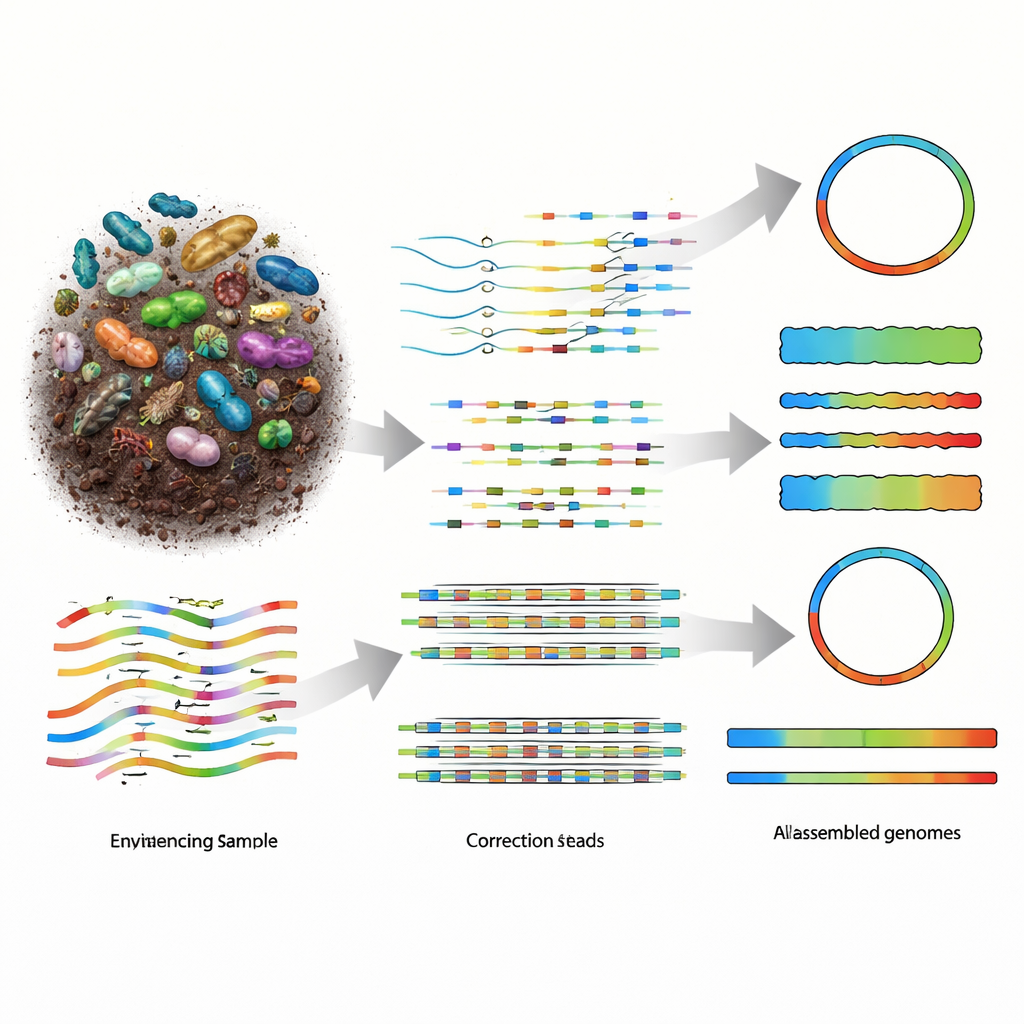
Warum das Wiederaufbauen von Genomen aus der Natur so schwierig ist
Metagenomik funktioniert, indem alle DNA‑Moleküle einer Probe in Bruchstücke zerlegt, diese Fragmente sequenziert und dann Software verwendet wird, um sie zu den Genomen der in der Probe vorhandenen Organismen zusammenzusetzen. Ältere Short‑Read‑Technologien lieferten viele kleine Stücke, doch die daraus resultierenden Puzzles waren stark fragmentiert, besonders in artenreichen Gemeinschaften wie der Bodenmikrobiota. Long‑Read‑Sequenzierplattformen, darunter PacBio HiFi und Oxford Nanopore Technologies (ONT), erzeugen viel längere DNA‑Stücke, was die Rekonstruktion erleichtern sollte. PacBios Reads sind extrem genau, aber teurer, während ONT‑Geräte günstiger und hochgradig portabel sind, historisch jedoch rauschärmere Daten lieferten. Als sich die ONT‑Chemie auf etwa einen Fehler pro hundert Basen verbesserte, bestanden Bedarf und Chancen für Assembler, diese neue Balance aus Länge, Genauigkeit und Kosten vollständig zu nutzen.
Von verrauschten Signalen zu sauberen Bausteinen
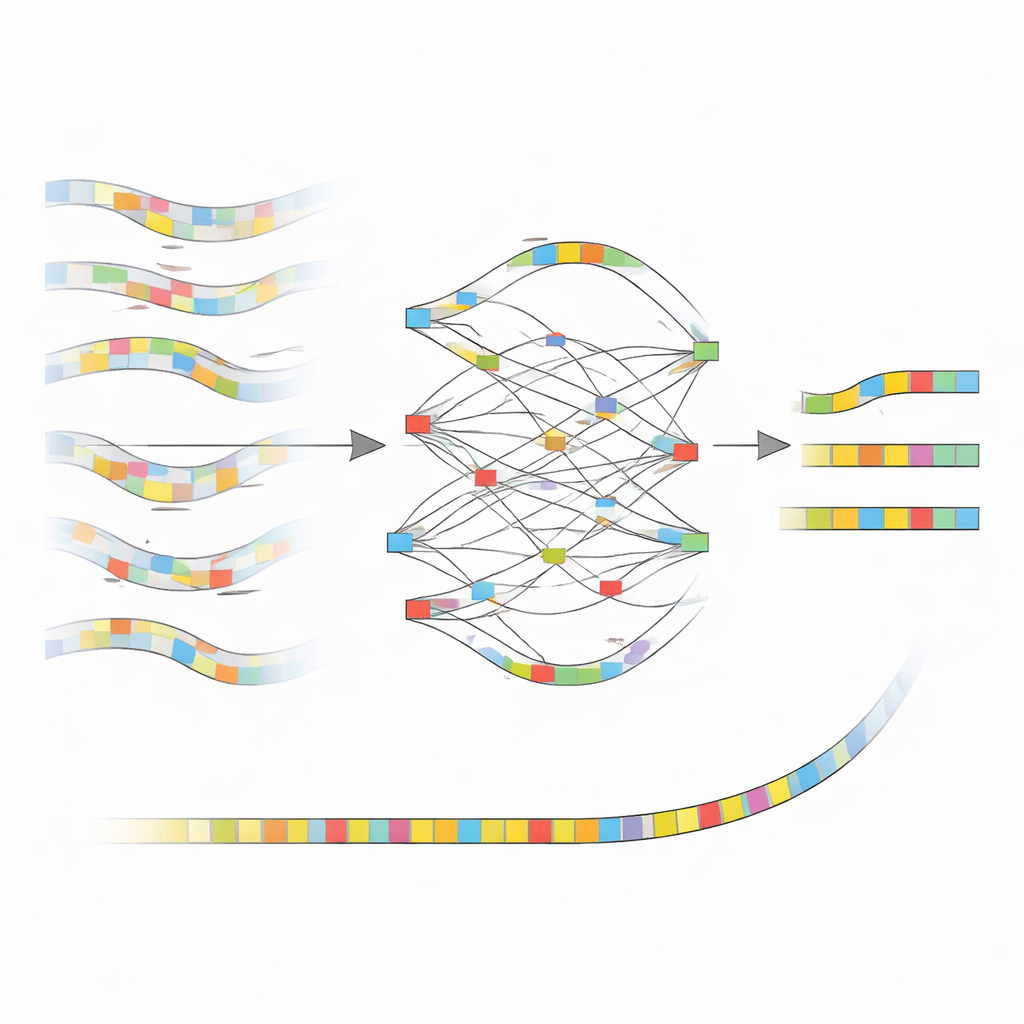
Die Kernidee von nanoMDBG ist, mit einer kompakten Skizze jedes DNA‑Fragments zu arbeiten statt mit jedem einzelnen Buchstaben. Die Methode wählt aus jedem Read eine dünn gestreute Menge kurzer DNA‑Muster, sogenannte Minimizer, und behandelt die geordnete Liste dieser Muster als leichtgewichtigen Fingerabdruck. Frühere Software derselben Gruppe, metaMDBG, nutzte solche Minimizer‑Fingerprints bereits, um sehr genaue PacBio‑Reads effizient zu assemblieren. Die verbleibenden Fehler in ONT‑Daten führten jedoch dazu, dass diese Fingerprints oft zerstört wurden, was Lücken und Fehlverknüpfungen zur Folge hatte. NanoMDBG begegnet dem, indem es ONT‑Reads zunächst in diesem reduzierten »Minimizer‑Raum« korrigiert. Für jeden Ziel‑Read rekrutiert der Algorithmus schnell eine Handvoll der ähnlichsten Reads mithilfe sehr spärlicher Fingerprints und prüft diese dann mit dichteren Fingerprints erneut, um falsche Übereinstimmungen von nicht verwandten Arten auszusortieren.
Wie die neue Methode das Bild bereinigt
Sobald nanoMDBG eine vertrauenswürdige Gruppe ähnlicher Fingerprints gesammelt hat, überlagert es diese, um einen einfachen Graphen zu bauen, der verfolgt, wo die Muster übereinstimmen, abweichen oder Einsätze und Löschungen zeigen. Statt jede Base zu betrachten, arbeitet es nur mit diesen ausgewählten Mustern und reduziert damit die Rechenlast erheblich. Der am stärksten unterstützte Pfad durch diesen Graphen wird zur Konsensus‑Fingerprint für den Read und glättet effektiv viele der ursprünglichen Sequenzierfehler aus. Alle korrigierten Fingerprints werden dann in den vorhandenen metaMDBG‑Assembler eingespeist, der sie zu längeren DNA‑Stücken zusammennäht und schließlich wieder in vollständige Sequenzen konvertiert, gefolgt von einem Polier‑Schritt zur Verfeinerung verbleibender kleiner Fehler.
nanoMDBG in realen Mikrobiomen getestet
Die Forschenden bewerteten nanoMDBG an mehreren Testfällen: einer definierten Mischung aus 21 bekannten Mikroben, einer menschlichen Darmprobe, einer Referenzmischung aus menschlichem Stuhlmaterial und einem sehr komplexen landwirtschaftlichen Boden. Sie verglichen die Leistung mit führenden Long‑Read‑Assemblern, namentlich metaFlye und dem früheren metaMDBG, und prüften, wie viele nahezu vollständige Genome — sogenannte metagenomisch zusammengesetzte Genome oder MAGs — sich rekonstruieren ließen und wie viele davon als einzelne, zusammenhängende Fragmente vorlagen. In allen drei realen Gemeinschaften erzeugte nanoMDBG deutlich mehr hochwertige MAGs als konkurrierende Werkzeuge und erheblich mehr vollständige Single‑Contig‑Genome. Im 400‑Milliarden‑Basen‑Bodendatensatz zum Beispiel rekonstruierte es 201 mehr nahezu vollständige Genome als metaMDBG und 144 mehr als metaFlye, während es nur einen Bruchteil des Arbeitsspeichers verbrauchte und in etwa sechs Tagen fertig war statt in fast einem Monat.
Teure Genauigkeit mit günstigeren Reads erreichen
Weil ONT‑ und PacBio‑Sequenzen auf denselben Proben mit vergleichbarer Tiefe erzeugt wurden, konnte das Team die Technologien direkt vergleichen. Für Darm‑ und standardisierte Stuhlgemeinschaften hatte PacBio HiFi weiterhin einen Vorsprung bei der Gesamtzahl höchstwertiger Genome, besonders bei höheren Sequenziertiefen. ONT‑Daten, assemblert mit nanoMDBG, kamen dem Ergebnis jedoch überraschend nahe und übertrafen HiFi in einigen Niedrig‑Tiefe‑Bedingungen sogar. Für die Bodenprobe, in der Tausende von Arten koexistieren, waren die Anzahlen nahezu vollständiger Genome von ONT und HiFi bei hoher Tiefe im Wesentlichen vergleichbar, wobei HiFi häufiger vollständig kontinuierliche Single‑Contig‑Genome erreichte. Detaillierte Fehleranalysen zeigten, dass nanoMDBG Fehlassemblierungen und Bereiche mit fehlender Abdeckung relativ gering hielt und mehr vollständige protein‑kodierende Gene bewahrte als konkurrierende ONT‑Assembler, besonders im anspruchsvollen Bodensatz.
Was das für die Erforschung unsichtbarer Ökosysteme bedeutet
Für Nicht‑Spezialisten lautet die Hauptbotschaft: Günstige, vor Ort einsetzbare DNA‑Sequenzierer können nun mikrobiellen Genomaufbau aus komplexen Umgebungen in einer Qualität rekonstruieren, die an größere, teurere Instrumente heranreicht. NanoMDBG erreicht dies, indem es die Daten geschickt in wiederverwendbare Muster vereinfacht, Fehler in dieser kompakten Darstellung korrigiert und dann Genome effizient aus den bereinigten Mustern zusammenbaut. Das macht es praktikabel, viele Proben zu untersuchen, mikrobielle Stämme über Personen oder Orte hinweg zu verfolgen und die enorme, größtenteils noch unerforschte Vielfalt des Lebens in Böden und anderen Habitaten zu erkunden — und das ohne Supercomputer‑große Ressourcen. Mit fortschreitender Verbesserung der Algorithmen werden solche Werkzeuge uns immer näher an routinemäßige, genomweite Karten ganzer mikrobieller Gemeinschaften bringen.
Zitation: Benoit, G., James, R., Raguideau, S. et al. High-quality metagenome assembly from nanopore reads with nanoMDBG. Nat Commun 17, 3556 (2026). https://doi.org/10.1038/s41467-026-69760-y
Schlüsselwörter: Metagenomik, Nanopore‑Sequenzierung, Genome Assemblierung, Mikrobiom, Bioinformatik