Clear Sky Science · en
High-quality metagenome assembly from nanopore reads with nanoMDBG
Reading Hidden Life in a Spoonful of Soil
Every pinch of soil or drop of gut fluid teems with thousands of microbial species, most of which cannot be grown in the lab. To understand what they are and what they do, scientists read their DNA directly from the environment, a field known as metagenomics. This article introduces nanoMDBG, a new computational method that turns raw signals from a portable DNA sequencer into high-quality draft genomes, opening the door to mapping complex microbial worlds faster, cheaper, and in far greater detail than before.
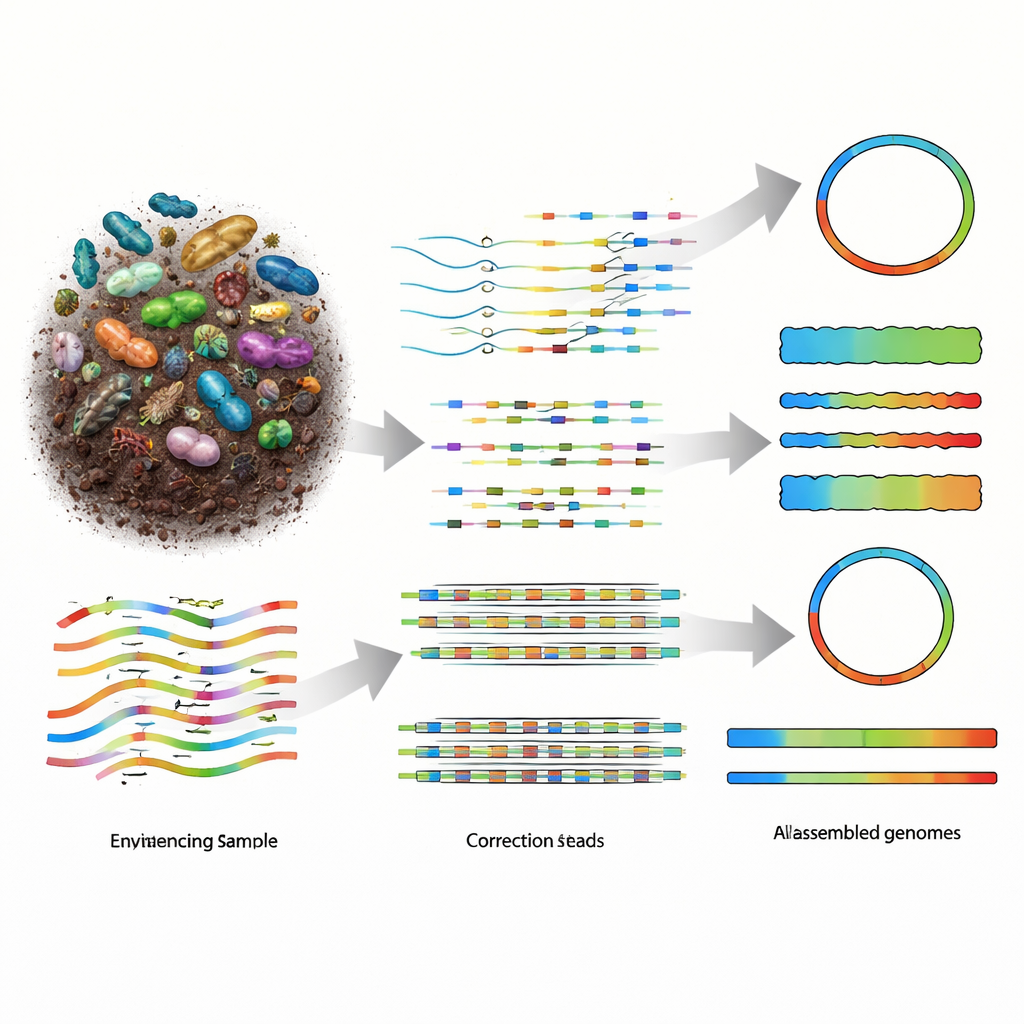
Why Rebuilding Genomes from Nature Is So Hard
Metagenomics works by shredding all the DNA in a sample into long fragments, sequencing those fragments, and then using software to reassemble them into the genomes of the organisms that were present. Older short-read technologies provided many small pieces, but the resulting puzzles were badly fragmented, especially in diverse communities such as soil. Long-read sequencing platforms, including PacBio HiFi and Oxford Nanopore Technologies (ONT), produce much longer pieces of DNA, which should make reconstruction easier. PacBio’s reads are extremely accurate but costlier, whereas ONT’s devices are more affordable and highly portable, but historically produced noisier data. As ONT chemistry improved to roughly one error in a hundred DNA letters, the field needed assemblers that could fully exploit this new balance of length, accuracy, and cost.
From Noisy Signals to Clean Building Blocks
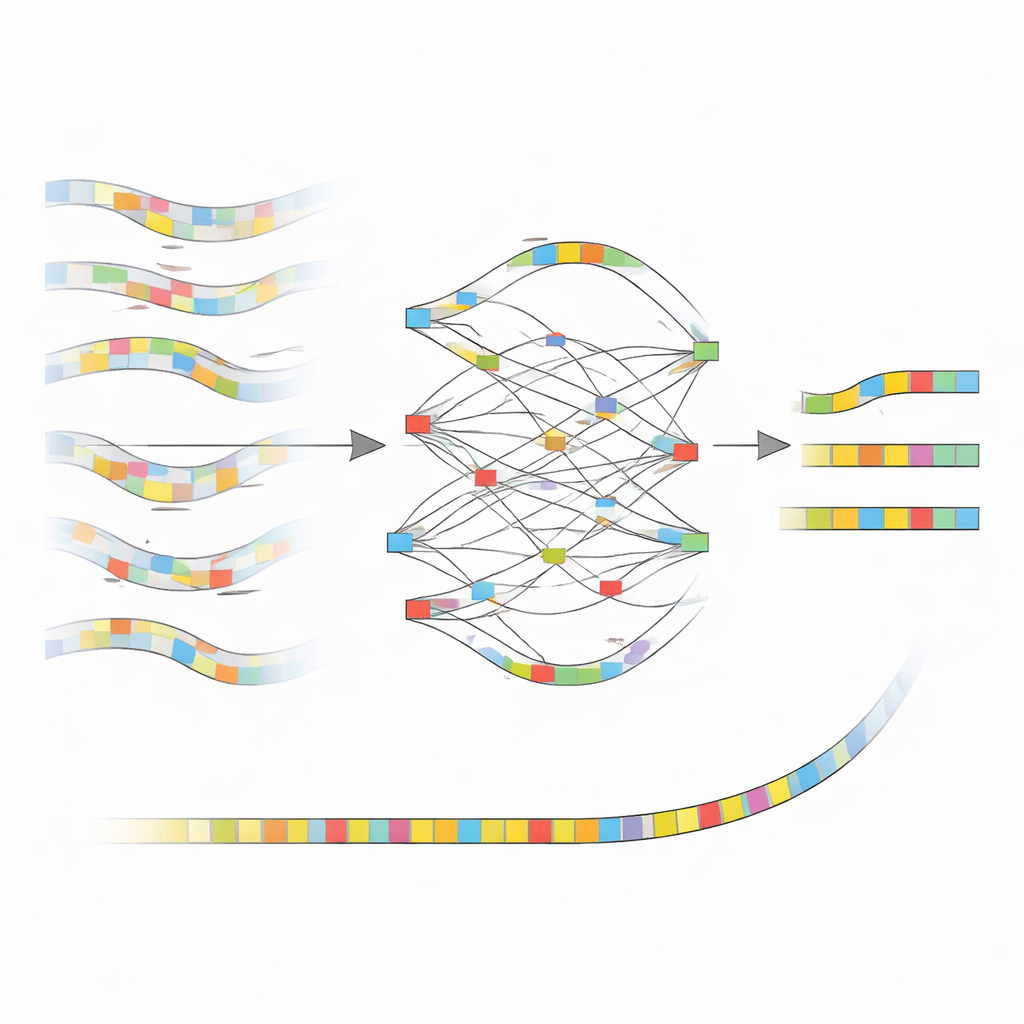
The core idea behind nanoMDBG is to work with a compact sketch of each DNA fragment instead of every single letter. The method selects a sparse set of short DNA patterns, called minimizers, from each read and treats the ordered list of these patterns as a lightweight fingerprint. Earlier software from the same group, metaMDBG, already used these minimizer fingerprints to assemble very accurate PacBio reads efficiently. However, the remaining errors in ONT data tended to break these fingerprints, leading to gaps and misjoins. NanoMDBG tackles this by first correcting ONT reads in this reduced “minimizer space.” For each target read, the algorithm quickly recruits a handful of the most similar reads using very sparse fingerprints, then re-examines them with denser fingerprints to weed out spurious matches from unrelated species.
How the New Method Cleans Up the Picture
Once nanoMDBG has gathered a trustworthy group of similar fingerprints, it overlays them to build a simple graph that tracks where the patterns agree, disagree, or show insertions and deletions. Instead of looking at every base, it works only with these selected patterns, greatly reducing the computing burden. The most strongly supported path through this graph becomes a consensus fingerprint for the read, effectively ironing out many of the original sequencing mistakes. All corrected fingerprints are then fed into the existing metaMDBG assembler, which stitches them into longer pieces of DNA and finally converts them back into full sequences, followed by a polishing step to refine remaining small errors.
Putting nanoMDBG to the Test in Real Microbiomes
The researchers evaluated nanoMDBG on several testbeds: a defined mixture of 21 known microbes, a human gut sample, a reference mixture of human fecal material, and a very complex agricultural soil. They compared its performance with leading long-read assemblers, notably metaFlye and the earlier metaMDBG, looking at how many near-complete genomes—known as metagenome-assembled genomes, or MAGs—they could recover, and how many of those were captured as single continuous pieces. Across all three real-world communities, nanoMDBG produced substantially more high-quality MAGs than competing tools, and many more complete, single-contig genomes. In the 400-billion-base soil dataset, for example, it recovered 201 more near-complete genomes than metaMDBG and 144 more than metaFlye, while using only a fraction of the memory and finishing in about six days instead of nearly a month.
Matching Costly Accuracy with Cheaper Reads
Because ONT and PacBio sequencers were run on the same samples at matched depths, the team could directly compare technologies. For gut and standardized fecal communities, PacBio HiFi still had an edge in the total number of top-quality genomes, especially at higher sequencing depths. Yet ONT data assembled with nanoMDBG came surprisingly close, and even outperformed HiFi in some low-depth conditions. For the soil sample, where thousands of species coexist, the numbers of near-complete genomes from ONT and HiFi were essentially comparable at high depth, though HiFi more often achieved fully continuous single-contig genomes. Detailed error analyses showed that nanoMDBG kept misassemblies and missing-coverage regions relatively low and preserved more full-length protein-coding genes than competing ONT assemblers, especially in the challenging soil dataset.
What This Means for Exploring Invisible Ecosystems
For non-specialists, the key message is that cheap, field-deployable DNA sequencers can now reconstruct microbial genomes from complex environments at a quality approaching that of larger, more expensive instruments. NanoMDBG accomplishes this by cleverly simplifying the data into reusable patterns, correcting errors in that compact representation, and then assembling genomes from the cleaned patterns with high efficiency. This makes it feasible to survey many samples, track microbial strains across people or locations, and explore the enormous, still mostly uncharted diversity of life in soils and other habitats, all without supercomputer-scale resources. As algorithms continue to improve, such tools will bring us ever closer to routine, genome-level maps of entire microbial communities.
Citation: Benoit, G., James, R., Raguideau, S. et al. High-quality metagenome assembly from nanopore reads with nanoMDBG. Nat Commun 17, 3556 (2026). https://doi.org/10.1038/s41467-026-69760-y
Keywords: metagenomics, nanopore sequencing, genome assembly, microbiome, bioinformatics