Clear Sky Science · it
Assemblaggio metagenomico di alta qualità da letture nanopore con nanoMDBG
Leggere la vita nascosta in un cucchiaino di suolo
Ogni pizzico di suolo o goccia di fluido intestinale è popolato da migliaia di specie microbiche, la maggior parte delle quali non può essere coltivata in laboratorio. Per capire chi sono e cosa fanno, gli scienziati leggono direttamente il loro DNA dall’ambiente, un campo noto come metagenomica. Questo articolo presenta nanoMDBG, un nuovo metodo computazionale che trasforma i segnali grezzi di un lettore di DNA portatile in bozze di genomi di alta qualità, aprendo la strada alla mappatura di mondi microbici complessi più rapidamente, a costi inferiori e con un livello di dettaglio molto maggiore rispetto al passato.
Perché ricostruire i genomi dalla natura è così difficile
La metagenomica funziona frammentando tutto il DNA in un campione in pezzi di varie lunghezze, sequenziando quei frammenti e poi usando software per riassemblarli nei genomi degli organismi presenti. Le tecnologie a lettura breve del passato fornivano molti piccoli pezzi, ma i puzzle risultanti erano fortemente frammentati, soprattutto in comunità molto diversificate come il suolo. Le piattaforme a lettura lunga, incluse PacBio HiFi e Oxford Nanopore Technologies (ONT), producono frammenti di DNA molto più lunghi, il che dovrebbe facilitare la ricostruzione. Le letture PacBio sono estremamente accurate ma più costose, mentre i dispositivi ONT sono più accessibili e portatili, ma storicamente generavano dati più rumorosi. Con il miglioramento della chimica ONT fino a circa un errore ogni cento basi, il campo aveva bisogno di assemblatori in grado di sfruttare appieno questo nuovo equilibrio tra lunghezza, accuratezza e costo.
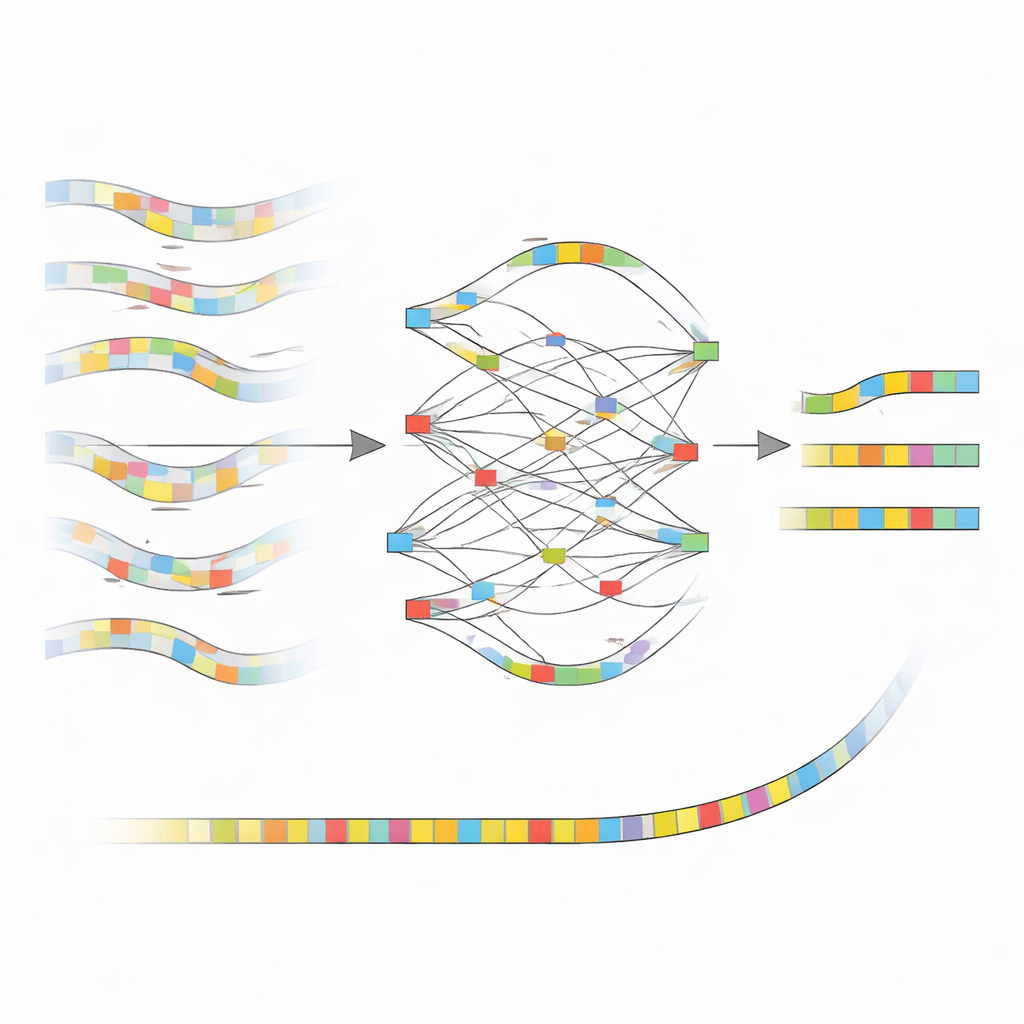
Dai segnali rumorosi ai mattoni puliti
L’idea centrale dietro nanoMDBG è lavorare con uno schizzo compatto di ciascun frammento di DNA invece che con ogni singola base. Il metodo seleziona un insieme sparso di brevi pattern di DNA, detti minimizer, da ogni lettura e considera la lista ordinata di questi pattern come un’impronta digitale leggera. Un software precedente dello stesso gruppo, metaMDBG, usava già queste impronte di minimizer per assemblare in modo efficiente letture PacBio molto accurate. Tuttavia, gli errori residui nei dati ONT tendevano a spezzare queste impronte, causando lacune e giunzioni errate. NanoMDBG risolve il problema correggendo prima le letture ONT in questo ridotto “spazio dei minimizer”. Per ogni lettura target, l’algoritmo recluta rapidamente un piccolo gruppo delle letture più simili usando impronte molto sparse, quindi le riesamina con impronte più dense per filtrare corrispondenze spurie provenienti da specie non correlate.
Come il nuovo metodo chiarisce il quadro
Una volta che nanoMDBG ha raccolto un gruppo affidabile di impronte simili, le sovrappone per costruire un grafo semplice che tiene traccia dei punti in cui i pattern concordano, divergono o mostrano inserzioni e delezioni. Invece di analizzare ogni base, lavora solo con questi pattern selezionati, riducendo notevolmente il carico computazionale. Il percorso maggiormente supportato attraverso questo grafo diventa un’impronta di consenso per la lettura, annullando di fatto molti degli errori di sequenziamento originali. Tutte le impronte corrette vengono poi fornite all’assemblatore metaMDBG esistente, che le cuce in pezzi di DNA più lunghi e infine le converte nuovamente in sequenze complete, seguite da una fase di polishing per affinare gli eventuali piccoli errori rimasti.
Mettere nanoMDBG alla prova in microbiomi reali
I ricercatori hanno valutato nanoMDBG su diversi casi di test: una miscela definita di 21 microrganismi noti, un campione di intestino umano, una miscela di riferimento di materiale fecale umano e un suolo agricolo molto complesso. Hanno confrontato le sue prestazioni con i principali assemblatori per letture lunghe, in particolare metaFlye e il precedente metaMDBG, esaminando quanti genomi quasi completi — noti come metagenome-assembled genomes, o MAG — riuscissero a ricostruire e quanti di questi fossero ottenuti come pezzi singoli continui. In tutte e tre le comunità reali, nanoMDBG ha prodotto sostanzialmente più MAG di alta qualità rispetto agli strumenti concorrenti, e molti più genomi completi in singoli contig. Nel dataset di suolo da 400 miliardi di basi, ad esempio, ha recuperato 201 genomi quasi completi in più rispetto a metaMDBG e 144 in più rispetto a metaFlye, usando solo una frazione della memoria e completando l’analisi in circa sei giorni invece che in quasi un mese.
Uguagliare l’accuratezza costosa con letture più economiche
Poiché i sequenziatori ONT e PacBio sono stati eseguiti sugli stessi campioni a profondità comparabili, il team ha potuto confrontare direttamente le tecnologie. Per le comunità intestinali e i campioni fecali standardizzati, PacBio HiFi manteneva ancora un vantaggio nel numero totale di genomi di massima qualità, specialmente ad alti livelli di sequenziamento. Tuttavia, i dati ONT assemblati con nanoMDBG si sono rivelati sorprendentemente vicini e in alcuni casi hanno superato HiFi in condizioni di bassa profondità. Per il campione di suolo, dove coesistono migliaia di specie, il numero di genomi quasi completi ottenuti con ONT e HiFi era essenzialmente comparabile ad alta profondità, sebbene HiFi riuscisse più spesso a ottenere genomi completamente continui in singoli contig. Analisi dettagliate degli errori hanno mostrato che nanoMDBG mantiene relativamente basse le misassemblaggi e le regioni a copertura mancante, preservando più geni codificanti proteine a lunghezza intera rispetto agli assemblatori ONT concorrenti, specialmente nel difficile dataset del suolo.
Cosa significa questo per l’esplorazione di ecosistemi invisibili
Per i non specialisti, il messaggio chiave è che sequenziatori di DNA economici e utilizzabili in campo possono ora ricostruire genomi microbici provenienti da ambienti complessi con una qualità prossima a quella di strumenti più grandi e costosi. NanoMDBG raggiunge questo obiettivo semplificando intelligentemente i dati in pattern riutilizzabili, correggendo gli errori in quella rappresentazione compatta e poi assemblando i genomi a partire dai pattern puliti con alta efficienza. Ciò rende praticabile l’analisi di molti campioni, il monitoraggio di ceppi microbici tra persone o luoghi e l’esplorazione dell’enorme e ancora per lo più inesplorata diversità di vita nei suoli e in altri habitat, il tutto senza risorse informatiche di scala supercomputer. Con il continuo miglioramento degli algoritmi, questi strumenti ci avvicineranno sempre più a mappe di comunità microbiche a livello genomico di routine.
Citazione: Benoit, G., James, R., Raguideau, S. et al. High-quality metagenome assembly from nanopore reads with nanoMDBG. Nat Commun 17, 3556 (2026). https://doi.org/10.1038/s41467-026-69760-y
Parole chiave: metagenomica, sequenziamento nanopore, assemblaggio del genoma, microbioma, bioinformatica