Clear Sky Science · pl
Wysokiej jakości montaż metagenomu z odczytów nanopore przy użyciu nanoMDBG
Odczytywanie ukrytego życia w łyżeczce gleby
Każdy szczypt gleb i każda kropla płynu jelitowego tętnią życiem — są w nich tysiące gatunków mikroorganizmów, z których większość nie daje się hodować w laboratorium. Aby dowiedzieć się, kim są i czym się zajmują, naukowcy czytają ich DNA bezpośrednio z próbek środowiskowych — to dziedzina zwana metagenomiką. W tym artykule przedstawiamy nanoMDBG, nową metodę obliczeniową, która przekształca surowe sygnały z przenośnego sekwenatora DNA w szkice genomów o wysokiej jakości, otwierając drogę do szybszego, tańszego i znacznie dokładniejszego mapowania złożonych światów mikrobiologicznych.
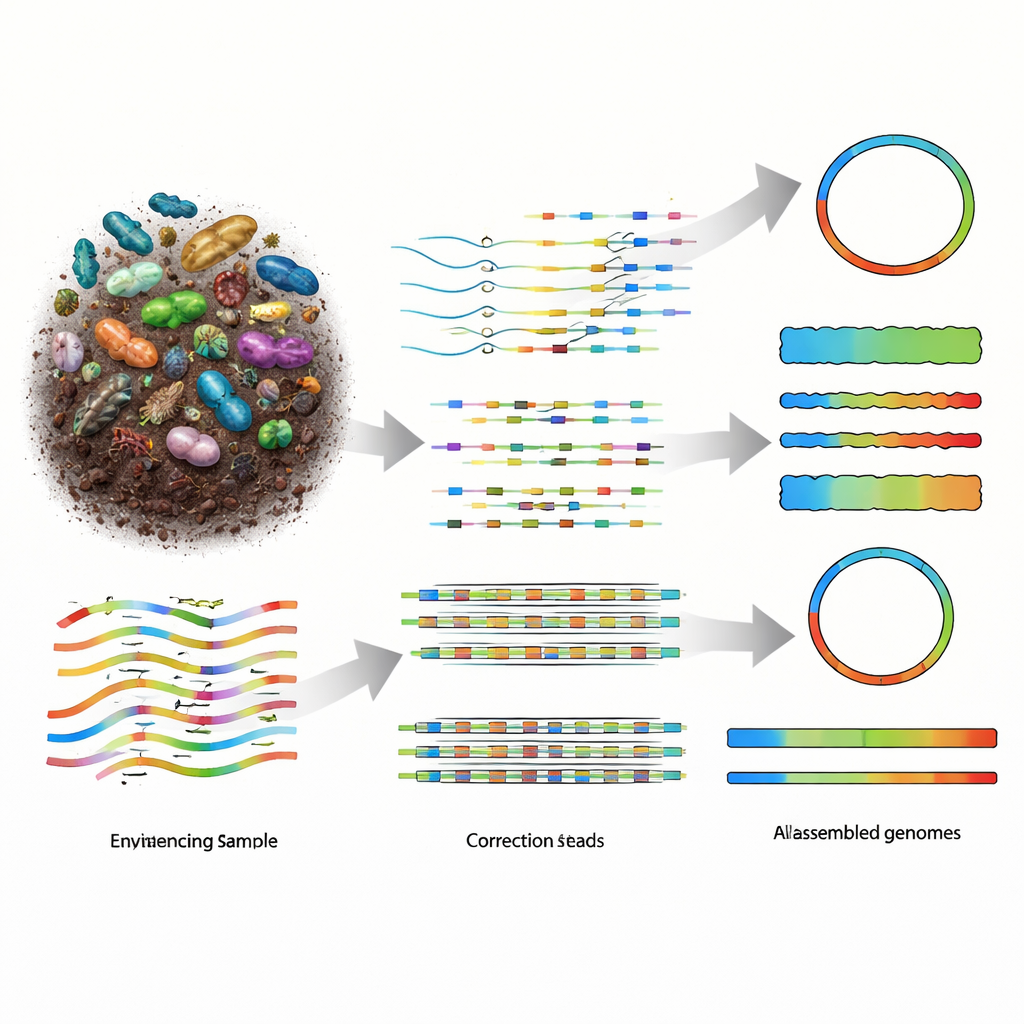
Dlaczego odtwarzanie genomów z natury jest tak trudne
Metagenomika polega na rozdrabnianiu całego DNA w próbce na fragmenty, sekwencjonowaniu tych fragmentów, a następnie użyciu oprogramowania do ponownego złożenia ich w genomy organizmów, które tam występowały. Starsze technologie krótkich odczytów dostarczały wielu małych kawałków, ale ułożone puzzle były mocno pofragmentowane, szczególnie w zróżnicowanych społecznościach, jak gleba. Platformy sekwencjonowania długich odczytów, w tym PacBio HiFi i Oxford Nanopore Technologies (ONT), generują znacznie dłuższe fragmenty, co teoretycznie powinno ułatwiać rekonstrukcję. Odczyty PacBio są wyjątkowo dokładne, lecz droższe, podczas gdy urządzenia ONT są tańsze i wysoce przenośne, ale historycznie dawały bardziej zaszumione dane. Gdy chemia ONT poprawiła się do poziomu około jednego błędu na sto zasad DNA, potrzeba było assemblerów potrafiących w pełni wykorzystać nowe połączenie długości, dokładności i kosztu.
Od zaszumionych sygnałów do czystych cegiełek
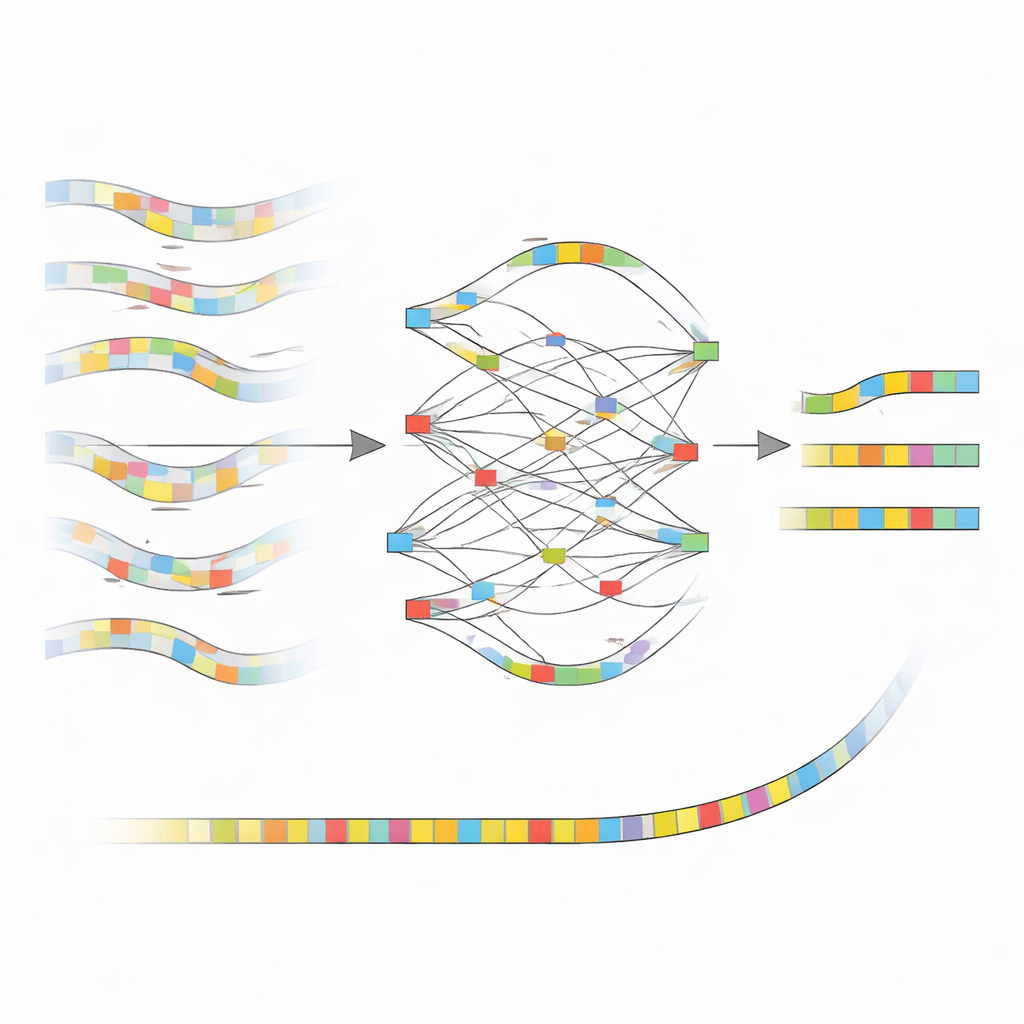
Kluczowy pomysł nanoMDBG polega na pracy z kompaktowym szkicem każdego fragmentu DNA zamiast z każdą pojedynczą zasadą. Metoda wybiera rzadką pulę krótkich wzorców DNA, zwanych minimizerami, z każdego odczytu i traktuje uporządkowaną listę tych wzorców jako lekkie odciski palców. Wcześniejsze oprogramowanie tej samej grupy, metaMDBG, już wykorzystywało takie minimizerowe odciski do wydajnego składania bardzo dokładnych odczytów PacBio. Jednak pozostałe błędy w danych ONT miały tendencję do łamania tych odcisków, prowadząc do braków i niepoprawnych złączy. NanoMDBG radzi sobie z tym, najpierw korygując odczyty ONT w zredukowanej „przestrzeni minimizerów”. Dla każdego odczytu docelowego algorytm szybko rekrutuje kilka najbardziej podobnych odczytów, używając bardzo rzadkich odcisków, po czym ponownie analizuje je gęstszymi odciskami, aby odsiać fałszywe dopasowania pochodzące od niepowiązanych gatunków.
Jak nowa metoda oczyszcza obraz
Gdy nanoMDBG zgromadzi zaufaną grupę podobnych odcisków, nakłada je na siebie, budując prosty graf śledzący miejsca, w których wzorce się zgadzają, różnią lub pokazują insercje i delecje. Zamiast analizować każdą zasadę, pracuje tylko z wybranymi wzorcami, co znacząco zmniejsza obciążenie obliczeniowe. Najmocniej wspierana ścieżka przez ten graf staje się konsensusem odcisku dla danego odczytu, skutecznie wygładzając wiele oryginalnych błędów sekwencjonowania. Wszystkie skorygowane odciski są następnie przekazywane do istniejącego assemblera metaMDBG, który składa je w dłuższe fragmenty DNA i ostatecznie odtwarza pełne sekwencje, po czym stosowany jest krok polerowania w celu poprawienia pozostałych drobnych błędów.
Testy nanoMDBG na prawdziwych mikrobiomach
Naukowcy ocenili nanoMDBG na kilku zestawach testowych: zdefiniowanej mieszance 21 znanych mikroorganizmów, próbce ludzkiego jelita, referencyjnej mieszance materiału kałowego oraz bardzo złożonej glebie rolniczej. Porównali jego wydajność z wiodącymi assemblerami odczytów długich, w szczególności metaFlye i wcześniejszym metaMDBG, analizując, ile niemal kompletnych genomów — znanych jako metagenome-assembled genomes (MAG) — udało się odzyskać oraz ile z nich odtworzono jako pojedyncze, ciągłe fragmenty. We wszystkich trzech społecznościach z rzeczywistego świata nanoMDBG wygenerował znacząco więcej MAG-ów wysokiej jakości niż konkurencyjne narzędzia i znacznie więcej kompletnych genomów w pojedynczych kontigach. W przypadku zestawu glebowego o objętości 400 miliardów zasad na przykład odzyskał o 201 więcej niemal kompletnych genomów niż metaMDBG i o 144 więcej niż metaFlye, przy użyciu jedynie ułamka pamięci i kończąc w około sześć dni zamiast prawie miesiąca.
Dopasowanie kosztownej dokładności tańszymi odczytami
Ponieważ sekwencjonery ONT i PacBio były uruchamiane na tych samych próbkach przy dopasowanej głębokości, zespół mógł bezpośrednio porównać technologie. Dla próbek jelitowych i zuniformizowanych społeczności kałowych PacBio HiFi wciąż miał przewagę w całkowitej liczbie genomów najwyższej jakości, zwłaszcza przy większych głębokościach sekwencjonowania. Niemniej jednak dane ONT złożone za pomocą nanoMDBG zbliżyły się zaskakująco blisko, a nawet przewyższyły HiFi w niektórych warunkach niskiej głębokości. Dla próbki gleby, gdzie współistnieje tysiące gatunków, liczby niemal kompletnych genomów z ONT i HiFi były w zasadzie porównywalne przy dużej głębokości, choć HiFi częściej osiągał w pełni ciągłe, pojedyncze kontigi. Szczegółowe analizy błędów wykazały, że nanoMDBG utrzymywał stosunkowo niskie wskaźniki błędnych złożeń i braków pokrycia oraz zachowywał więcej pełnych genów kodujących białka niż konkurencyjne assemblery ONT, zwłaszcza w wymagającym zestawie glebowym.
Co to oznacza dla eksploracji niewidzialnych ekosystemów
Dla osób spoza specjalizacji kluczowy wniosek jest taki, że tanie, przenośne sekwencjonery DNA potrafią teraz odbudowywać genomy mikroorganizmów z złożonych środowisk w jakości zbliżonej do większych, droższych instrumentów. NanoMDBG osiąga to poprzez sprytne uproszczenie danych do wielokrotnego użytku wzorców, korygowanie błędów w tej kompaktowej reprezentacji, a następnie składanie genomów z oczyszczonych wzorców z wysoką wydajnością. Umożliwia to badanie wielu próbek, śledzenie szczepów mikroorganizmów między ludźmi czy lokalizacjami oraz eksplorację ogromnej, wciąż w przeważającej mierze niezbadanej różnorodności życia w glebach i innych siedliskach — wszystko to bez zasobów na skalę superkomputerów. W miarę jak algorytmy będą się dalej poprawiać, takie narzędzia przybliżą nas do rutynowego, na poziomie genomu mapowania całych społeczności mikrobiologicznych.
Cytowanie: Benoit, G., James, R., Raguideau, S. et al. High-quality metagenome assembly from nanopore reads with nanoMDBG. Nat Commun 17, 3556 (2026). https://doi.org/10.1038/s41467-026-69760-y
Słowa kluczowe: metagenomika, sekwencjonowanie nanopore, składanie genomu, mikrobiom, bioinformatyka