Clear Sky Science · pt
De dados à descoberta: A ascensão de modelos preditivos baseados em teoria da informação no desenvolvimento de medicamentos
Por que descobrir medicamentos mais rápido importa
Muitas doenças graves ainda não têm tratamentos eficazes e, mesmo quando medicamentos promissores são encontrados, a jornada da ideia até as prateleiras da farmácia é longa e cara. Este artigo explora como modelos computacionais mais inteligentes podem vasculhar grandes coleções de dados químicos e biológicos para identificar alguns candidatos a fármaco de forma mais rápida e confiável. Ao tomar emprestado conceitos da teoria da informação — a matemática de quanto podemos aprender a partir dos dados — os autores mostram uma forma de estreitar a busca por novos medicamentos e compreender melhor o que torna uma molécula provável de funcionar no organismo.
Do teste e erro ao design orientado por dados
A descoberta tradicional de medicamentos dependia de uma mistura de palpites informados, triagens laboratoriais e, por vezes, acidentes felizes como a descoberta da penicilina. Hoje, pesquisadores podem testar milhões de compostos em computadores antes de tocar num tubo de ensaio. Ferramentas de triagem virtual classificam moléculas segundo seu comportamento biológico previsto, ajudando cientistas a focar nas mais promissoras. No entanto, muitas ferramentas existentes tratam cada molécula de forma isolada ou fornecem apenas estimativas grosseiras de probabilidade, e frequentemente têm dificuldade em capturar como o contexto biológico do mundo real — por exemplo, como um fármaco se desloca pelo corpo — molda sucesso ou fracasso.
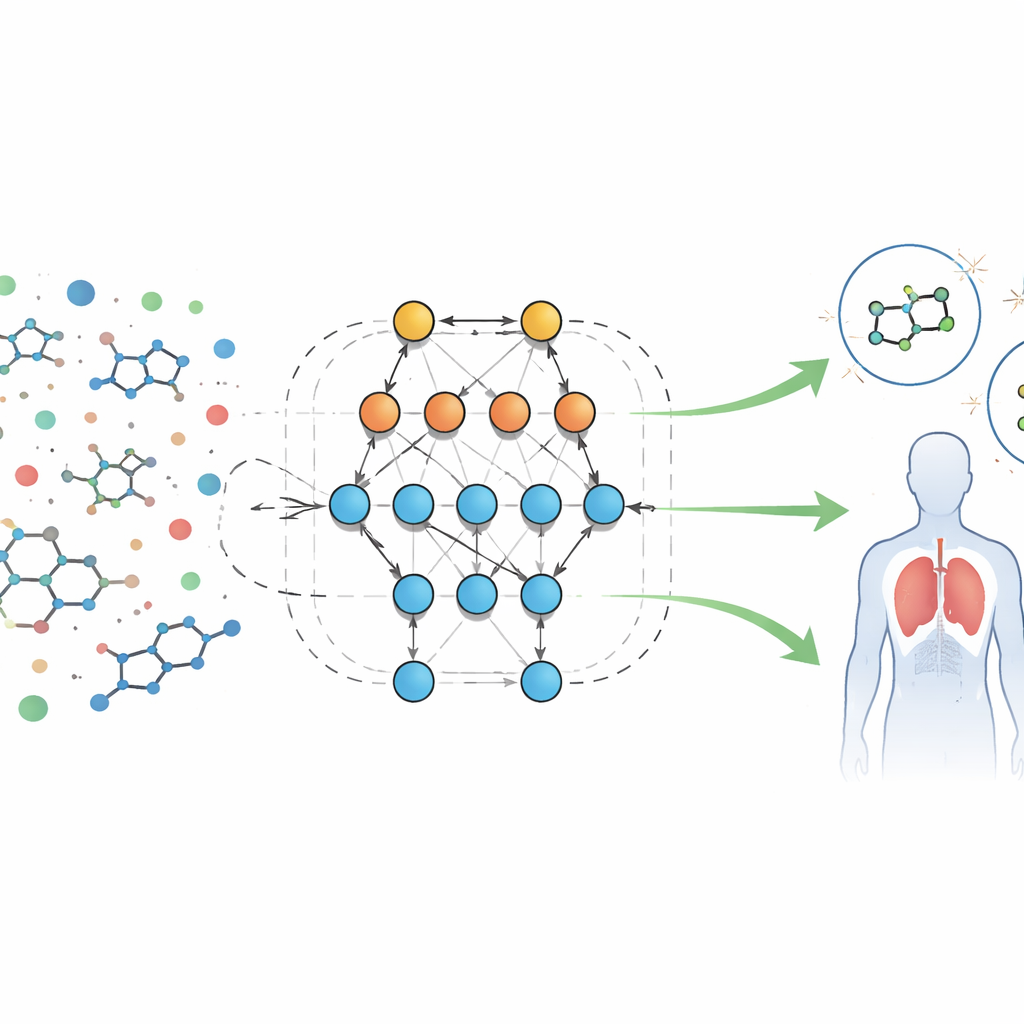
Uma nova forma de ler impressões biológicas
Os autores analisam um grande conjunto de dados públicos de resultados de triagem biológica para moléculas direcionadas a um receptor peptídico formil, uma proteína envolvida em inflamação e defesas imunes. Cada amostra vem com dezenas de características mensuráveis, ou “descritores”, como tamanho molecular, solubilidade em lipídios ou água, capacidade de atravessar a barreira hematoencefálica e aptidão para formar ligações de hidrogênio. Em vez de escrever equações fixas sobre como essas características devem se comportar, a equipe usa um sistema automatizado chamado Eidos, que constrói modelos preditivos baseados em teoria da informação diretamente a partir dos dados. Esses modelos, referidos como análise ASC (sistema-automatizado-cognitivo), aprendem como combinações de características se relacionam com o fato de uma amostra se comportar como ativa (potencialmente útil) ou inativa em testes biológicos.
Limpeza dos dados e escolha do que importa
Dados de triagem do mundo real são ruidosos: medições podem ser inconsistentes e algumas amostras podem não se encaixar em nenhum padrão claro. O sistema Eidos primeiro filtra esses “artefatos”, removendo mais de mil entradas questionáveis e mantendo pouco mais de duas mil amostras confiáveis. Em seguida, examina mais de 300 características para ver quais realmente ajudam a distinguir amostras ativas de inativas. Usando conceitos da teoria da informação, cada característica é pontuada conforme quanto reduz a incerteza sobre o resultado. A análise revela que apenas uma minoria de características carrega a maior parte da informação útil, o que significa que os pesquisadores podem ignorar com segurança muitas medições e ainda reter quase todo o poder preditivo. Esse corte torna os modelos mais simples, mais fáceis de interpretar e mais rápidos de executar.
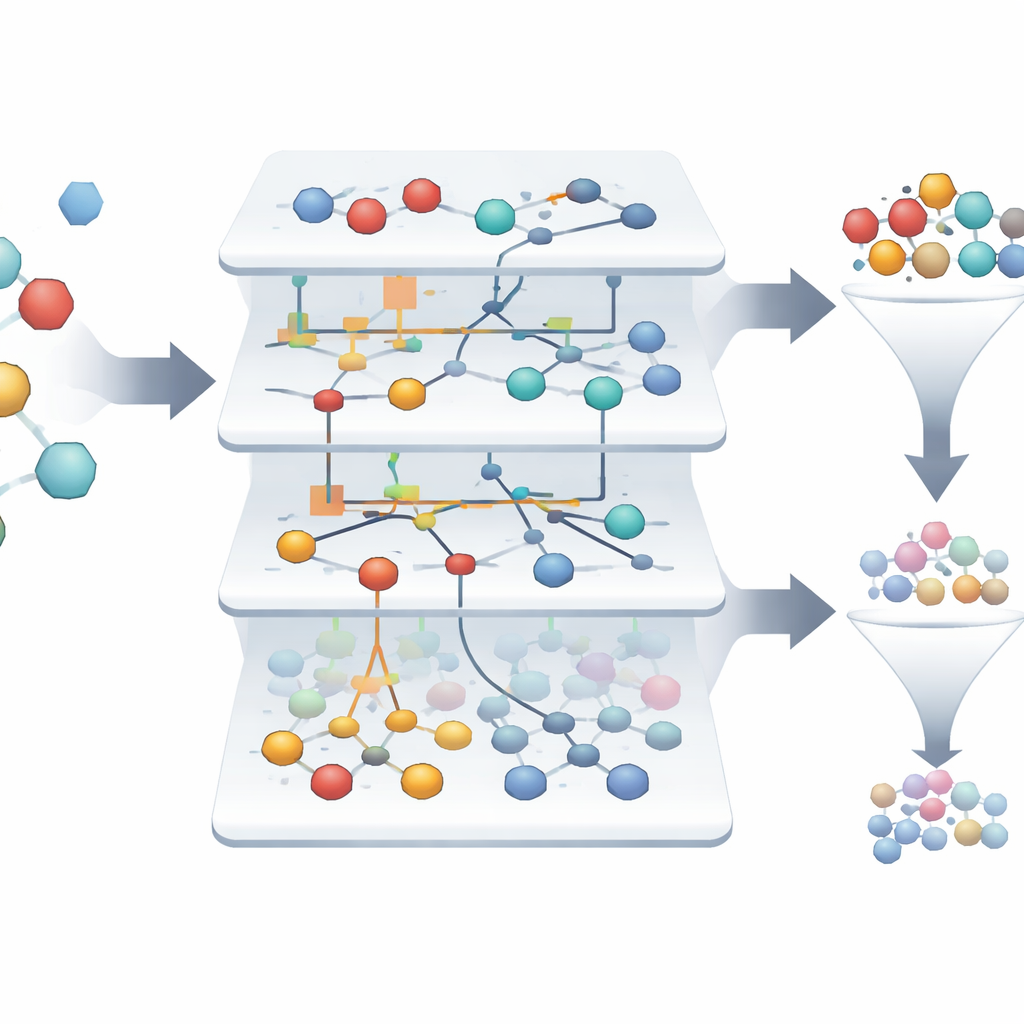
Encontrando vencedores raros num mar de falhas
No conjunto de dados estudado, apenas cerca de 1,4% das moléculas são realmente ativas, tornando desafiador identificar os poucos vencedores entre milhares de fracassos. Os modelos baseados em ASC constroem automaticamente “retratos informacionais” que mostram o quanto cada característica e combinação de características empurra uma amostra na direção de ser ativa ou inativa. De mais de três mil amostras originais, o sistema destaca apenas duas que se sobressaem como candidatas altamente confiáveis para fármacos que visam o receptor peptídico formil, com confiabilidade do modelo aproximando 99,9% em testes retrospectivos. Diagramas de rede visuais mostram quais características moleculares mais fortemente sustentam um estado ativo, oferecendo aos cientistas um mapa interpretável do que dirige um comportamento promissor.
Como essa abordagem se compara e o que vem a seguir
Os autores comparam seu método com ferramentas populares de predição em estágio inicial, como pkCSM, SwissADME e ADMETlab, que estimam como um fármaco é absorvido, distribuído, metabolizado e excretado. Enquanto esses sistemas dependem principalmente de regras predefinidas ou aprendizado de máquina de uso geral, a estrutura ASC mede explicitamente quanto cada característica contribui para o conhecimento obtido sobre comportamento parecido com fármaco e pode simular mudanças no contexto biológico. Ao mesmo tempo, o estudo aponta limites: o conjunto de dados é relativamente pequeno e fortemente desbalanceado, e o método até agora foi aplicado a apenas um receptor. Os autores sugerem que versões futuras poderiam combinar modelos ASC com aprendizado profundo e expandir para múltiplos alvos.
O que isso significa para medicamentos futuros
Na prática, este trabalho mostra que modelos ricos em informação podem transformar dados de triagem confusos em previsões claras e testáveis sobre quais moléculas merecem atenção adicional. Ao limpar automaticamente os dados, ranquear a importância das características e destacar compostos raros, porém promissores, a abordagem pode reduzir o tempo e o custo necessários para chegar ao laboratório e, eventualmente, à clínica. Embora não substitua estudos em animais ou ensaios em humanos, atua como um filtro e guia poderosos, ajudando cientistas a passar de dados brutos a potenciais terapias com mais eficiência e maior confiança.
Citação: Saied, H., Alfahad, O., Aljaffer, A.A. et al. From data to discovery: The rise of information-theoretic predictive models in drug development. Sci Rep 16, 12857 (2026). https://doi.org/10.1038/s41598-026-45644-5
Palavras-chave: descoberta de fármacos, triagem virtual, modelagem preditiva, dados de bioensaios, receptor peptídico formil