Clear Sky Science · en
From data to discovery: The rise of information-theoretic predictive models in drug development
Why faster drug discovery matters
Many serious illnesses still lack effective treatments, and even when promising medicines are found, the journey from idea to pharmacy shelf is long and expensive. This paper explores how smarter computer models can sift through huge collections of chemical and biological data to pinpoint a few promising drug candidates more quickly and reliably. By borrowing ideas from information theory—the mathematics of how much we can learn from data—the authors show a way to narrow the search for new medicines and better understand what makes a molecule likely to work in the body.
From trial and error to data-driven design
Traditional drug discovery has relied on a mix of educated guesswork, lab screening, and, at times, lucky accidents such as the discovery of penicillin. Today, researchers can test millions of compounds in computers before ever touching a test tube. Virtual screening tools classify molecules according to their predicted biological behavior, helping scientists focus on the most promising ones. However, many existing tools either treat each molecule in isolation or provide only rough probability estimates, and they often struggle to capture how real-world biological context—such as how a drug moves through the body—shapes success or failure.
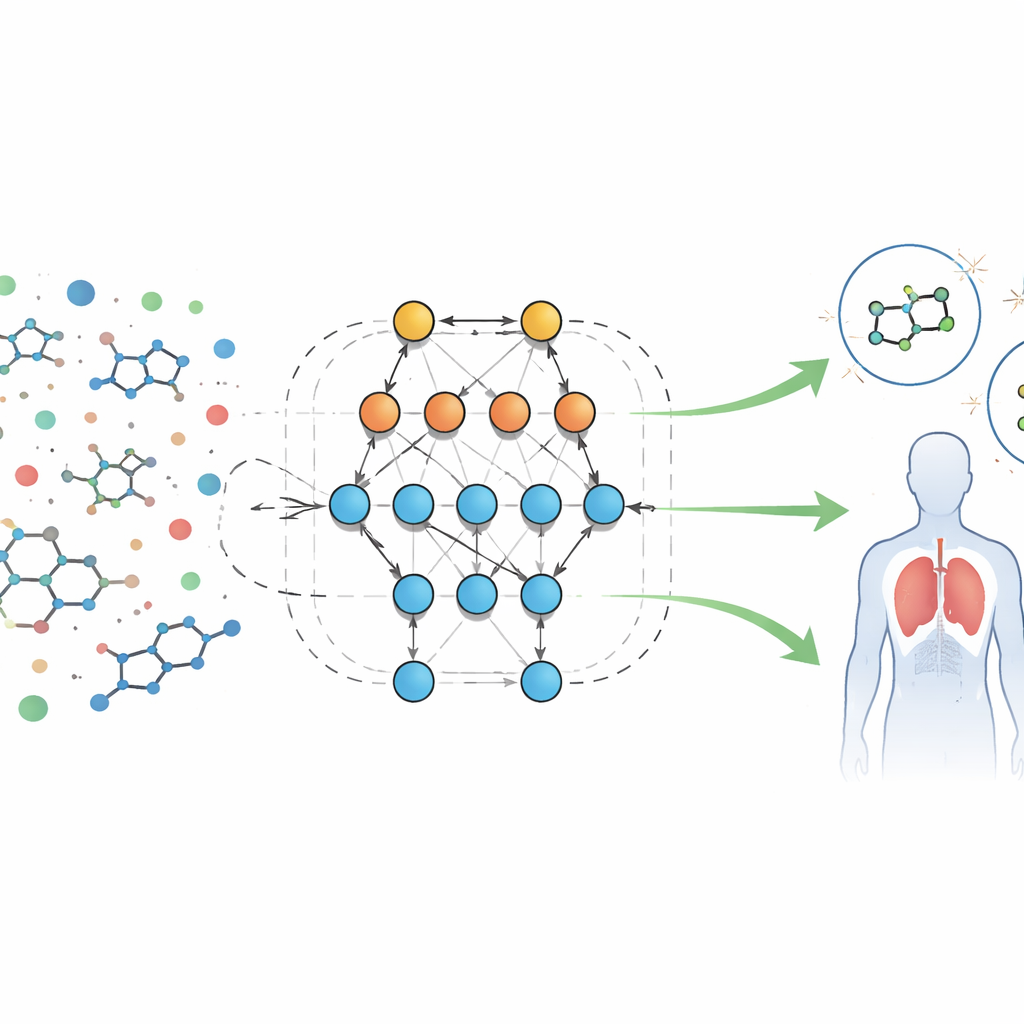
A new way to read biological fingerprints
The authors analyze a large public dataset of biological screening results for molecules aimed at a formyl peptide receptor, a protein involved in inflammation and immune defenses. Each sample comes with dozens of measurable features, or “descriptors,” such as molecular size, how easily it dissolves in fat or water, ability to cross the blood–brain barrier, and capacity to form hydrogen bonds. Instead of writing fixed equations for how these features should behave, the team uses an automated system called Eidos, which builds information-theoretic predictive models directly from the data. These models, referred to as ASC (automated system-cognitive) analysis, learn how combinations of features are linked to whether a sample behaves as active (potentially useful) or inactive in biological tests.
Cleaning the data and choosing what matters
Real-world screening data are noisy: measurements may be inconsistent, and some samples may not fit any clear pattern. The Eidos system first filters out these “artifacts,” removing over a thousand questionable entries and keeping just over two thousand reliable samples. It then examines more than 300 features to see which ones actually help distinguish active from inactive samples. Using concepts from information theory, each feature is scored by how much it reduces uncertainty about the outcome. The analysis reveals that only a minority of features carry most of the useful information, meaning researchers can safely ignore many measurements and still retain almost all predictive power. This trimming makes models simpler, easier to interpret, and faster to run.
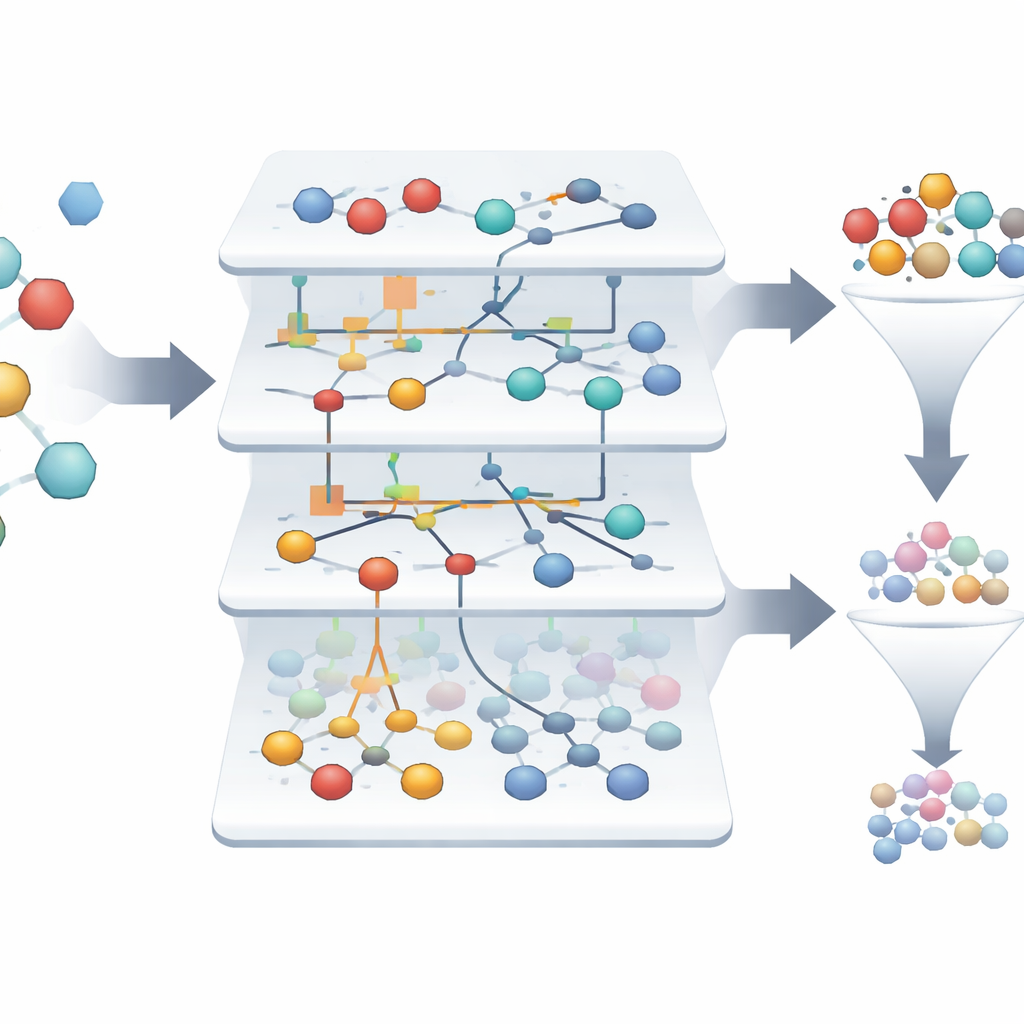
Finding rare winners in a sea of failures
In the dataset studied, only about 1.4% of the molecules are truly active, making it challenging to spot the few winners among thousands of failures. The ASC-based models automatically build “information portraits” that show how strongly each feature and feature combination nudges a sample toward being active or inactive. Out of more than three thousand original samples, the system highlights just two that stand out as highly reliable candidates for drugs targeting the formyl peptide receptor, with model reliability approaching 99.9% on retrospective tests. Visual network diagrams show which molecular characteristics most strongly support an active state, giving scientists an interpretable map of what drives promising behavior.
How this approach stacks up and what comes next
The authors compare their method with popular early-stage prediction tools such as pkCSM, SwissADME, and ADMETlab, which estimate how a drug is absorbed, distributed, broken down, and excreted. While those systems rely mainly on predefined rules or general-purpose machine learning, the ASC framework explicitly measures how much each feature contributes to the knowledge gained about drug-like behavior and can simulate changes in biological context. At the same time, the study notes limits: the dataset is relatively small and heavily imbalanced, and the method has so far been applied to only one receptor. The authors suggest that future versions could combine ASC models with deep learning and expand to multiple targets.
What it means for future medicines
In practical terms, this work shows that information-rich models can turn messy screening data into clear, testable predictions about which molecules deserve further attention. By automatically cleaning data, ranking the importance of features, and spotlighting rare yet promising compounds, the approach can reduce the time and cost needed to reach the lab and, eventually, the clinic. While it does not replace animal studies or human trials, it acts as a powerful filter and guide, helping scientists move from raw data to potential therapies more efficiently and with greater confidence.
Citation: Saied, H., Alfahad, O., Aljaffer, A.A. et al. From data to discovery: The rise of information-theoretic predictive models in drug development. Sci Rep 16, 12857 (2026). https://doi.org/10.1038/s41598-026-45644-5
Keywords: drug discovery, virtual screening, predictive modeling, bioassay data, formyl peptide receptor