Clear Sky Science · pl
Od danych do odkrycia: Wzrost modeli predykcyjnych opartych na teorii informacji w tworzeniu leków
Dlaczego szybsze odkrywanie leków ma znaczenie
Wiele poważnych chorób wciąż nie ma skutecznych terapii, a nawet gdy odkryje się obiecujące leki, droga od pomysłu do półki aptecznej jest długa i kosztowna. Artykuł ten bada, jak inteligentniejsze modele komputerowe mogą przesiać ogromne zbiory danych chemicznych i biologicznych, aby szybciej i pewniej wskazać kilka obiecujących kandydatów. Czerpiąc z teorii informacji — matematyki badającej, ile można dowiedzieć się z danych — autorzy pokazują sposób zawężania poszukiwań nowych leków i lepszego rozumienia, co sprawia, że cząsteczka ma większe szanse zadziałać w organizmie.
Od prób i błędów do projektowania opartego na danych
Tradycyjne odkrywanie leków opierało się na mieszance przemyślanych przypuszczeń, badań przesiewowych w laboratorium i czasem szczęśliwych przypadkach, takich jak odkrycie penicyliny. Dziś badacze mogą testować miliony związków w komputerze, zanim dotkną probówki. Narzędzia do wirtualnego screeningu klasyfikują cząsteczki według przewidywanego zachowania biologicznego, pomagając koncentrować się na najbardziej obiecujących. Jednak wiele istniejących narzędzi traktuje każdą cząsteczkę oddzielnie albo daje jedynie przybliżone estymaty prawdopodobieństwa, a także często ma trudności z uchwyceniem, jak rzeczywisty kontekst biologiczny — na przykład jak lek przemieszcza się w organizmie — wpływa na sukces lub porażkę.
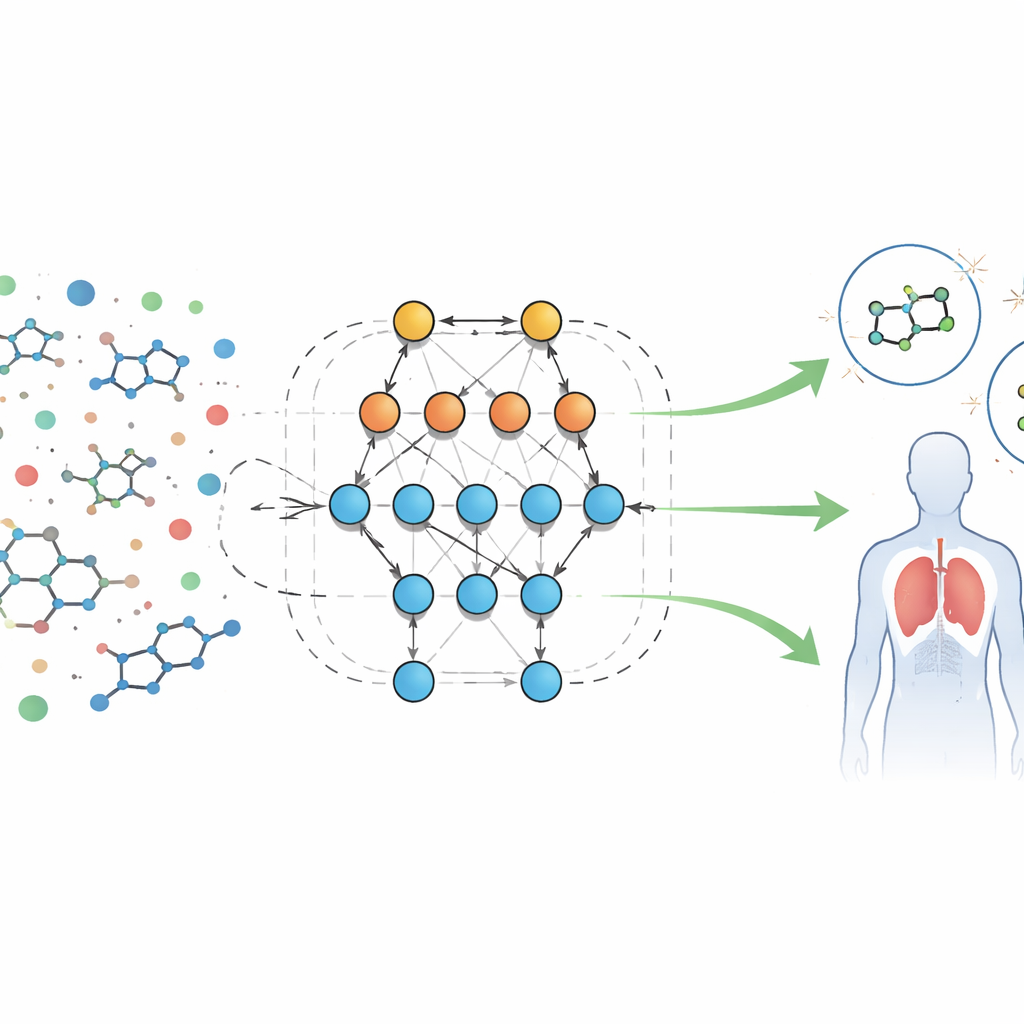
Nowy sposób odczytywania biologicznych odcisków palców
Autorzy analizują duży publiczny zbiór wyników badań przesiewowych dotyczących cząsteczek ukierunkowanych na receptor peptydu formylowego, białko zaangażowane w stany zapalne i obronę immunologiczną. Każdy próbka zawiera dziesiątki mierzalnych cech, czyli „deskryptorów”, takich jak rozmiar cząsteczki, rozpuszczalność w tłuszczach lub wodzie, zdolność przejścia przez barierę krew–mózg czy tworzenia wiązań wodorowych. Zamiast pisać stałe równania opisujące zachowanie tych cech, zespół używa zautomatyzowanego systemu o nazwie Eidos, który buduje predykcyjne modele oparte na teorii informacji bezpośrednio z danych. Modele te, określane jako analiza ASC (automated system-cognitive), uczą się, jak kombinacje cech korelują z tym, czy próbka zachowuje się jako aktywna (potencjalnie użyteczna) czy nieaktywna w testach biologicznych.
Oczyszczanie danych i wybór tego, co ważne
Dane przesiewowe z rzeczywistego świata są zaszumione: pomiary mogą być niespójne, a niektóre próbki nie pasują do żadnego wyraźnego wzorca. System Eidos najpierw filtruje te „artefakty”, usuwając ponad tysiąc wątpliwych wpisów i pozostawiając nieco ponad dwa tysiące wiarygodnych próbek. Następnie przegląda ponad 300 cech, aby ustalić, które z nich rzeczywiście pomagają odróżnić próbki aktywne od nieaktywnych. Korzystając z pojęć z teorii informacji, każda cecha otrzymuje ocenę według tego, o ile redukuje niepewność co do wyniku. Analiza ujawnia, że tylko mniejszość cech niesie większość użytecznych informacji, co oznacza, że badacze mogą bezpiecznie zignorować wiele pomiarów i wciąż zachować niemal całą moc predykcyjną. To przycinanie upraszcza modele, ułatwia ich interpretację i przyspiesza obliczenia.
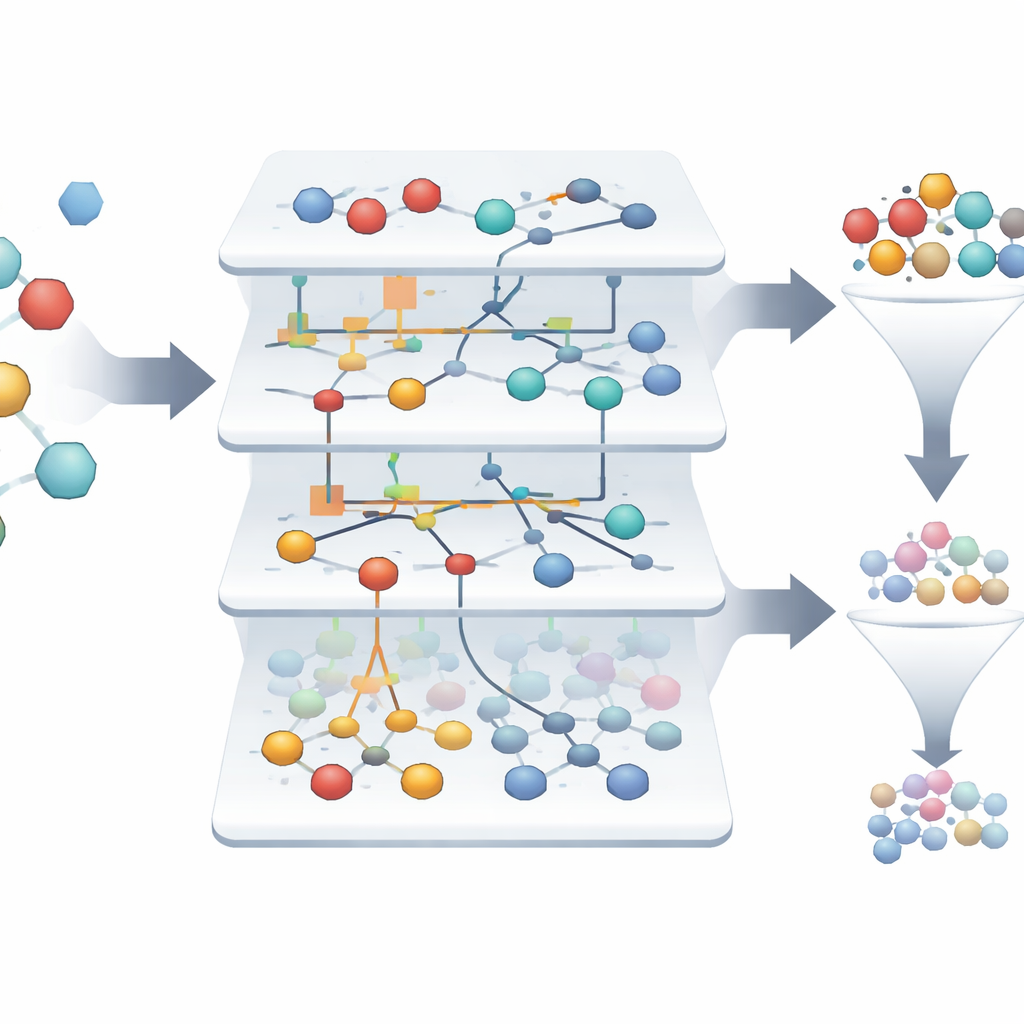
Znalezienie rzadkich zwycięzców w morzu porażek
W badanym zbiorze danych jedynie około 1,4% cząsteczek jest naprawdę aktywnych, co utrudnia wyłowienie kilku zwycięzców spośród tysięcy porażek. Modele oparte na ASC automatycznie tworzą „portrety informacyjne”, które pokazują, jak silnie każda cecha i kombinacja cech przesuwa próbkę w stronę stanu aktywnego lub nieaktywnego. Z ponad trzech tysięcy oryginalnych próbek system wyróżnia tylko dwie, które wybiegają jako wysoce wiarygodni kandydaci na leki skierowane przeciwko receptorowi peptydu formylowego, przy wiarygodności modelu zbliżającej się do 99,9% w testach retrospektywnych. Wizualne diagramy sieci pokazują, które cechy molekularne najsilniej wspierają stan aktywny, dając naukowcom interpretowalną mapę czynników warunkujących obiecujące zachowanie.
Jak to podejście wypada i co dalej
Autorzy porównują swoją metodę z popularnymi narzędziami do przewidywań we wczesnym etapie, takimi jak pkCSM, SwissADME i ADMETlab, które szacują, jak lek jest wchłaniany, rozprowadzany, metabolizowany i wydalany. Podczas gdy te systemy opierają się głównie na z góry określonych regułach lub ogólnych metodach uczenia maszynowego, ramy ASC mierzą wprost, ile każda cecha wnosi do wiedzy o zachowaniu podobnym do leku i potrafią symulować zmiany w kontekście biologicznym. Równocześnie badanie wskazuje ograniczenia: zbiór danych jest stosunkowo mały i silnie niezrównoważony, a metoda dotychczas zastosowana tylko do jednego receptora. Autorzy sugerują, że przyszłe wersje mogłyby łączyć modele ASC z uczeniem głębokim i rozszerzyć zastosowanie na wiele celów.
Co to oznacza dla przyszłych leków
W praktyce praca ta pokazuje, że modele bogate w informację mogą przekształcić nieuporządkowane dane przesiewowe w jasne, testowalne przewidywania dotyczące tego, które cząsteczki zasługują na dalsze zainteresowanie. Poprzez automatyczne oczyszczanie danych, rankingowanie ważności cech i wyłanianie rzadkich, ale obiecujących związków, podejście to może skrócić czas i obniżyć koszty potrzebne na dotarcie do laboratorium, a w końcu do kliniki. Choć nie zastępuje badań na zwierzętach ani badań klinicznych na ludziach, działa jako potężny filtr i przewodnik, pomagając naukowcom przejść od surowych danych do potencjalnych terapii szybciej i z większą pewnością.
Cytowanie: Saied, H., Alfahad, O., Aljaffer, A.A. et al. From data to discovery: The rise of information-theoretic predictive models in drug development. Sci Rep 16, 12857 (2026). https://doi.org/10.1038/s41598-026-45644-5
Słowa kluczowe: odkrywanie leków, screening wirtualny, modelowanie predykcyjne, dane z bioassay, receptor peptydu formylowego