Clear Sky Science · de
Von Daten zur Entdeckung: Der Aufstieg informationstheoretischer Vorhersagemodelle in der Arzneimittelentwicklung
Warum schnellere Wirkstofffindung wichtig ist
Viele schwere Erkrankungen haben noch keine wirksamen Behandlungen, und selbst wenn vielversprechende Wirkstoffe gefunden werden, ist der Weg von der Idee bis zur Apotheke lang und kostspielig. Dieser Beitrag untersucht, wie intelligentere Computermodelle große Sammlungen chemischer und biologischer Daten durchsieben können, um schneller und zuverlässiger einige wenige vielversprechende Wirkstoffkandidaten zu identifizieren. Indem sie Konzepte aus der Informationstheorie — der Mathematik darüber, wie viel wir aus Daten lernen können — nutzen, zeigen die Autoren einen Weg auf, die Suche nach neuen Medikamenten einzugrenzen und besser zu verstehen, was eine Molekülwirkung im Körper wahrscheinlich macht.
Vom Versuch‑und‑Irrtum zur datengetriebenen Gestaltung
Die traditionelle Wirkstoffforschung beruhte auf einer Mischung aus fundierten Vermutungen, Labortests und gelegentlich glücklichen Zufällen wie der Entdeckung des Penicillins. Heute können Forschende Millionen von Verbindungen am Computer testen, bevor sie ein Reagenzglas anfassen. Werkzeuge für das virtuelle Screening klassifizieren Moleküle nach ihrem vorhergesagten biologischen Verhalten und helfen so, die Aufmerksamkeit auf die vielversprechendsten Kandidaten zu lenken. Viele vorhandene Werkzeuge behandeln jedoch jedes Molekül isoliert oder liefern nur grobe Wahrscheinlichkeitsaussagen und tun sich schwer damit, wie biologische Kontextfaktoren — etwa die Verteilung eines Wirkstoffs im Körper — Erfolg oder Misserfolg beeinflussen.
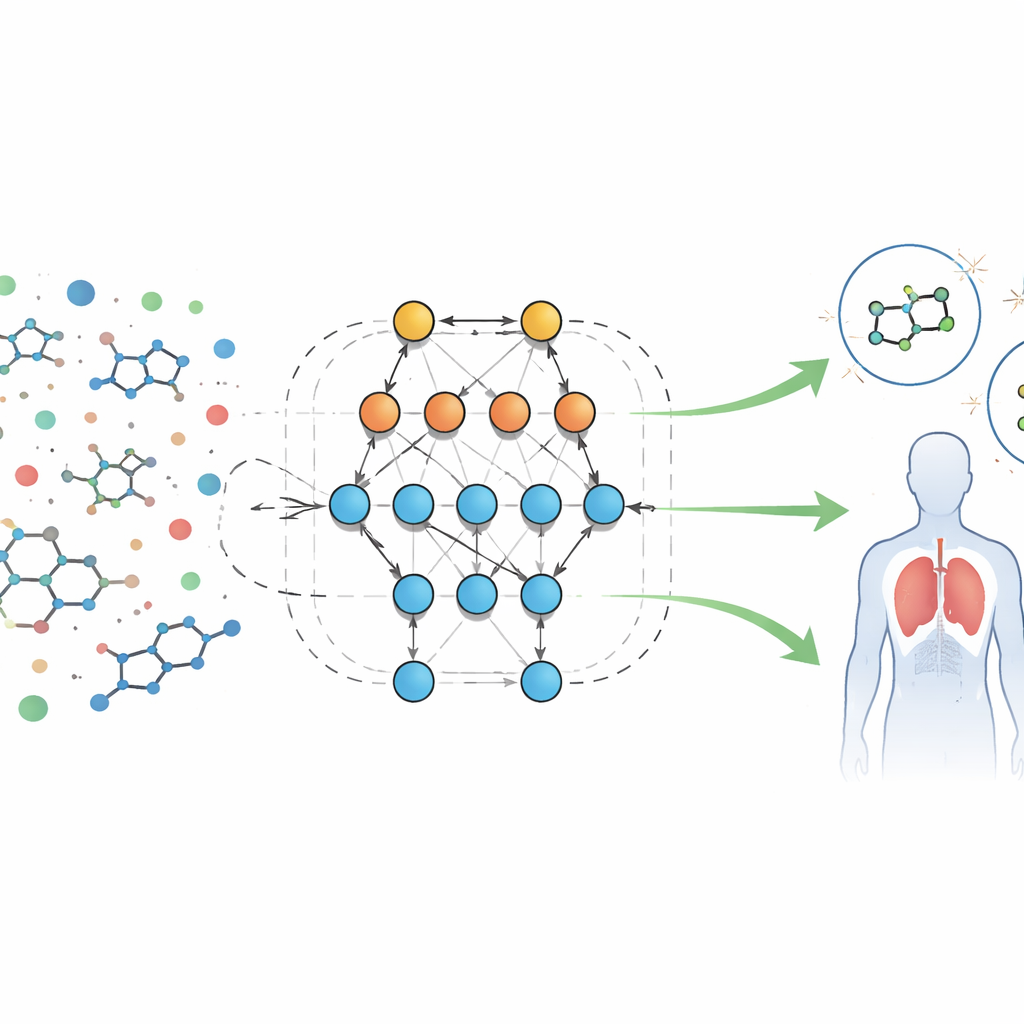
Eine neue Art, biologische Fingerabdrücke zu lesen
Die Autoren analysieren einen großen öffentlichen Datensatz mit biologischen Screening-Ergebnissen für Moleküle, die auf einen Formylpeptid-Rezeptor zielen, ein Protein, das an Entzündungs- und Immunreaktionen beteiligt ist. Jede Probe enthält Dutzende messbarer Eigenschaften oder „Deskriptoren“, wie Molekülgröße, Lipophilie (Löslichkeit in Fett vs. Wasser), Fähigkeit, die Blut‑Hirn-Schranke zu überwinden, und das Potenzial zur Ausbildung von Wasserstoffbrücken. Statt feste Gleichungen dafür vorzugeben, wie sich diese Merkmale verhalten sollten, verwendet das Team ein automatisiertes System namens Eidos, das informationstheoretische Vorhersagemodelle direkt aus den Daten aufbaut. Diese Modelle, als ASC-Analyse (automated system-cognitive) bezeichnet, lernen, wie Kombinationen von Merkmalen mit dem Verhalten einer Probe in biologischen Tests — aktiv (potenziell nützlich) oder inaktiv — verknüpft sind.
Daten säubern und das Wichtige auswählen
Reale Screening-Daten sind rauschbehaftet: Messungen können inkonsistent sein, und manche Proben passen in kein klares Muster. Das Eidos-System filtert zunächst solche „Artefakte“ heraus, entfernt über tausend fragwürdige Einträge und behält etwas mehr als zweitausend verlässliche Proben. Anschließend untersucht es mehr als 300 Merkmale, um zu ermitteln, welche tatsächlich dabei helfen, aktive von inaktiven Proben zu unterscheiden. Mit Konzepten aus der Informationstheorie wird jedes Merkmal danach bewertet, wie sehr es die Unsicherheit über das Ergebnis reduziert. Die Analyse zeigt, dass nur eine Minderheit der Merkmale den Großteil der nützlichen Information trägt, sodass Forschende viele Messungen sicher vernachlässigen können, ohne nahezu die gesamte Vorhersagekraft zu verlieren. Dieses Beschneiden macht die Modelle einfacher, besser interpretierbar und schneller lauffähig.
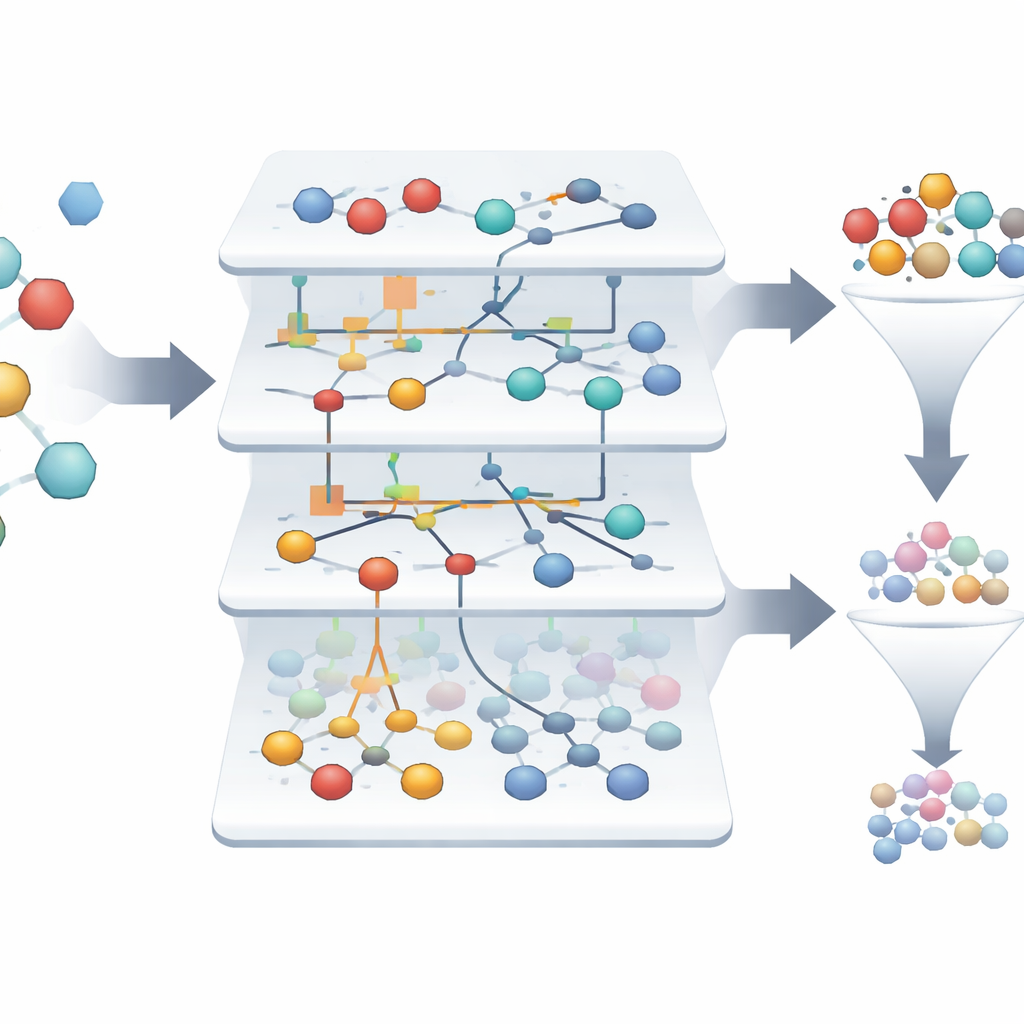
Seltene Gewinner in einem Meer von Misserfolgen finden
Im untersuchten Datensatz sind nur etwa 1,4 % der Moleküle tatsächlich aktiv, was es schwierig macht, die wenigen Gewinner unter Tausenden von Ausfällen zu erkennen. Die ASC-basierten Modelle bauen automatisch „Informationsporträts“ auf, die zeigen, wie stark jedes Merkmal und jede Merkmalkombination eine Probe in Richtung aktiv oder inaktiv schiebt. Von mehr als dreitausend ursprünglichen Proben hebt das System nur zwei hervor, die als hochzuverlässige Kandidaten für Wirkstoffe gegen den Formylpeptid-Rezeptor gelten, mit einer Modellzuverlässigkeit, die in retrospektiven Tests nahe 99,9 % liegt. Visuelle Netzwerkdiagramme zeigen, welche molekularen Eigenschaften einen aktiven Zustand am stärksten stützen und geben Forschenden eine interpretierbare Karte der Treiber vielversprechenden Verhaltens.
Wie sich der Ansatz einordnet und was als Nächstes kommt
Die Autoren vergleichen ihre Methode mit bekannten frühen Vorhersagewerkzeugen wie pkCSM, SwissADME und ADMETlab, die abschätzen, wie ein Wirkstoff resorbiert, verteilt, metabolisiert und ausgeschieden wird. Während diese Systeme hauptsächlich auf vordefinierten Regeln oder allgemeinen Machine‑Learning‑Ansätzen beruhen, misst das ASC‑Framework explizit, wie viel jedes Merkmal zum gewonnenen Wissen über wirkstoffähnliches Verhalten beiträgt, und kann Veränderungen im biologischen Kontext simulieren. Zugleich weisen die Autoren auf Grenzen hin: Der Datensatz ist relativ klein und stark unausgewogen, und die Methode wurde bisher nur auf einen Rezeptor angewandt. Sie schlagen vor, künftige Versionen mit Deep Learning zu kombinieren und auf mehrere Ziele auszuweiten.
Was das für künftige Medikamente bedeutet
Praktisch zeigt diese Arbeit, dass informationsreiche Modelle unstrukturierte Screening‑Daten in klare, prüfbare Vorhersagen darüber verwandeln können, welche Moleküle weitere Aufmerksamkeit verdienen. Durch automatisches Säubern der Daten, Rangfolge der Merkmalsbedeutung und Hervorhebung seltener, aber vielversprechender Verbindungen kann der Ansatz Zeit und Kosten bis zum Labor und letztlich zur Klinik reduzieren. Er ersetzt zwar nicht Tierversuche oder klinische Studien, dient aber als kraftvoller Filter und Leitfaden, der Forschenden hilft, effizienter und mit größerer Zuversicht von Rohdaten zu potenziellen Therapien zu gelangen.
Zitation: Saied, H., Alfahad, O., Aljaffer, A.A. et al. From data to discovery: The rise of information-theoretic predictive models in drug development. Sci Rep 16, 12857 (2026). https://doi.org/10.1038/s41598-026-45644-5
Schlüsselwörter: Wirkstoffforschung, virtuelles Screening, vorhersagende Modellierung, Bioassay-Daten, Formylpeptid-Rezeptor