Clear Sky Science · fr
Des données à la découverte : l’essor des modèles prédictifs informationnels en développement de médicaments
Pourquoi accélérer la découverte de médicaments est important
De nombreuses maladies graves restent sans traitements efficaces, et même lorsque des médicaments prometteurs sont identifiés, le parcours de l’idée jusqu’à la mise en rayon est long et coûteux. Cet article examine comment des modèles informatiques plus intelligents peuvent trier d’immenses collections de données chimiques et biologiques pour repérer plus rapidement et de manière plus fiable quelques candidats-médicaments prometteurs. En empruntant des concepts à la théorie de l’information — les mathématiques de ce que l’on peut apprendre à partir des données — les auteurs proposent une façon de restreindre la recherche de nouveaux médicaments et de mieux comprendre ce qui rend une molécule susceptible d’agir dans l’organisme.
Du tâtonnement à la conception guidée par les données
La découverte traditionnelle de médicaments s’est appuyée sur un mélange d’hypothèses éclairées, de criblages en laboratoire et, parfois, de coups de chance comme la découverte de la pénicilline. Aujourd’hui, les chercheurs peuvent tester des millions de composés par ordinateur avant d’avoir touché un tube à essai. Les outils de dépistage virtuel classent les molécules selon leur comportement biologique prédit, aidant les scientifiques à se concentrer sur les plus prometteuses. Cependant, beaucoup d’outils existants traitent chaque molécule de façon isolée ou ne fournissent que des estimations probabilistes approximatives, et ils peinent souvent à saisir comment le contexte biologique réel — par exemple la manière dont un médicament se déplace dans l’organisme — façonne le succès ou l’échec.
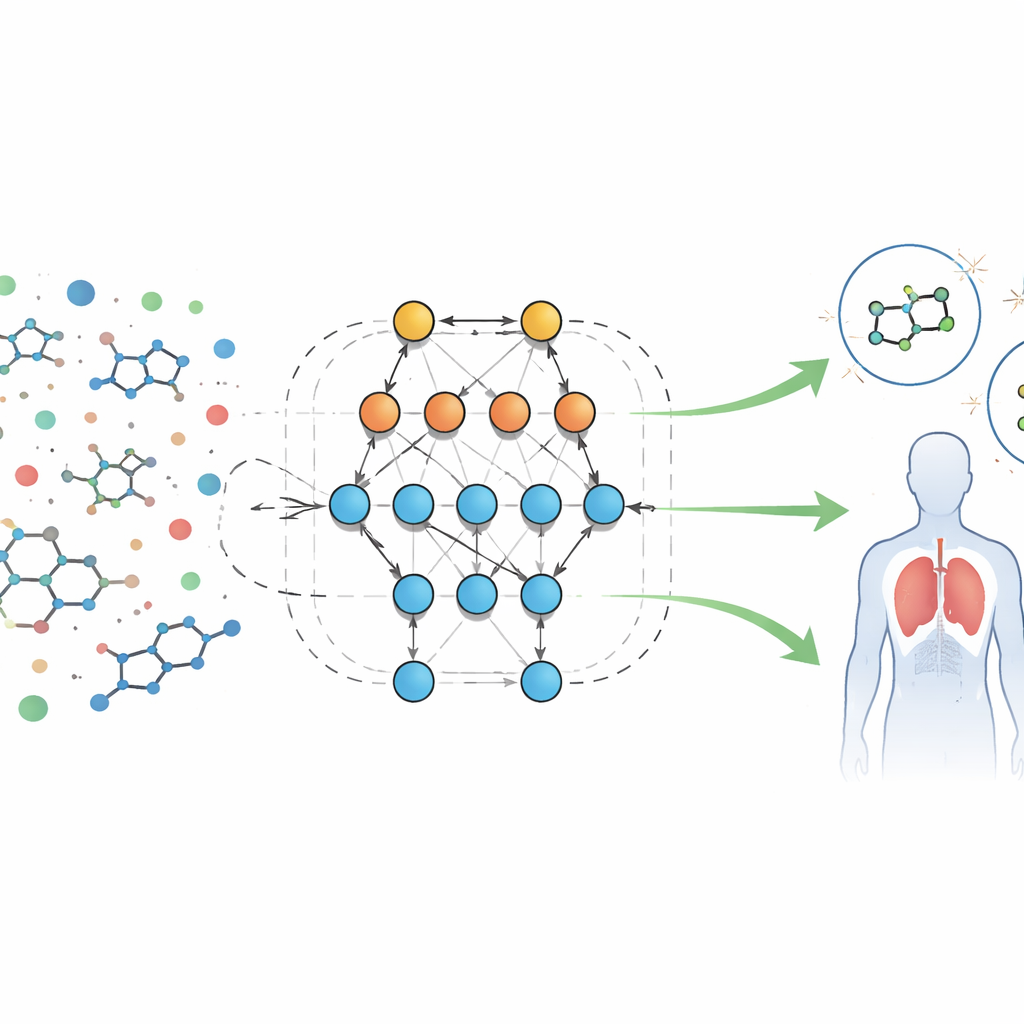
Une nouvelle façon de lire les empreintes biologiques
Les auteurs analysent un large jeu de données public de résultats de criblage biologique pour des molécules ciblant un récepteur peptidique formylé, une protéine impliquée dans l’inflammation et les défenses immunitaires. Chaque échantillon s’accompagne de dizaines de caractéristiques mesurables, ou « descripteurs », tels que la taille moléculaire, la solubilité dans les graisses ou l’eau, la capacité à traverser la barrière hémato-encéphalique et la propension à former des liaisons hydrogène. Plutôt que d’écrire des équations fixes sur le comportement attendu de ces caractéristiques, l’équipe utilise un système automatisé nommé Eidos, qui construit des modèles prédictifs basés sur la théorie de l’information directement à partir des données. Ces modèles, désignés sous le nom d’analyse ASC (automated system-cognitive), apprennent comment des combinaisons de caractéristiques se lient au fait qu’un échantillon soit actif (potentiellement utile) ou inactif dans les tests biologiques.
Nettoyer les données et choisir ce qui compte
Les données de criblage du monde réel sont bruyantes : les mesures peuvent être incohérentes et certains échantillons ne correspondent à aucun schéma clair. Le système Eidos filtre d’abord ces « artefacts », en retirant plus d’un millier d’entrées douteuses et en ne conservant qu’un peu plus de deux mille échantillons fiables. Il examine ensuite plus de 300 caractéristiques pour voir lesquelles aident réellement à distinguer les échantillons actifs des inactifs. En utilisant des concepts de la théorie de l’information, chaque caractéristique se voit attribuer un score mesurant la réduction de l’incertitude sur l’issue. L’analyse révèle que seule une minorité de descripteurs porte la majeure partie de l’information utile, ce qui signifie que les chercheurs peuvent ignorer en toute sécurité de nombreuses mesures tout en conservant presque toute la puissance prédictive. Cet élagage rend les modèles plus simples, plus faciles à interpréter et plus rapides à exécuter.
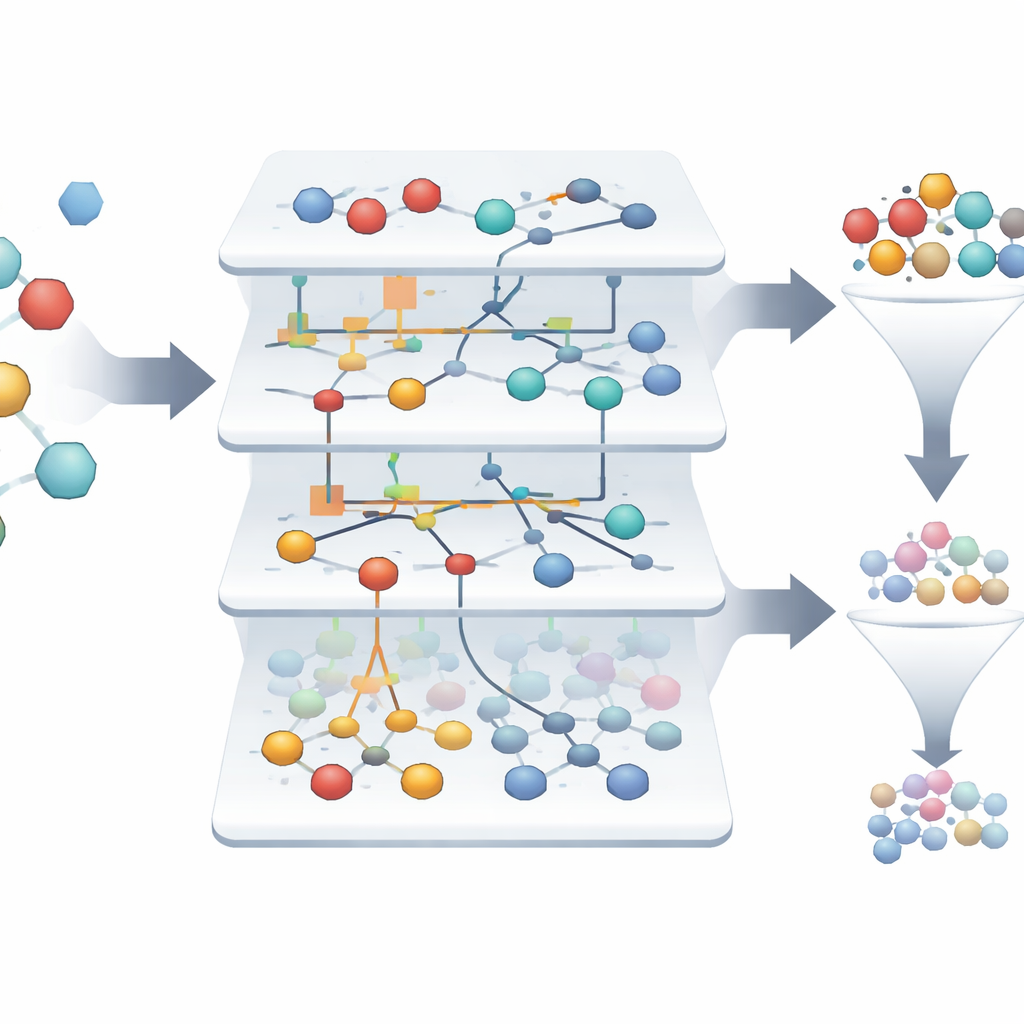
Trouver de rares gagnants dans une mer d’échecs
Dans le jeu de données étudié, seulement environ 1,4 % des molécules sont réellement actives, ce qui rend difficile la détection des quelques gagnants parmi des milliers d’échecs. Les modèles basés sur l’ASC construisent automatiquement des « portraits informationnels » montrant dans quelle mesure chaque caractéristique et chaque combinaison de caractéristiques incline un échantillon vers l’état actif ou inactif. Sur plus de trois mille échantillons initiaux, le système met en évidence seulement deux qui se démarquent comme des candidats hautement fiables pour cibler le récepteur peptidique formylé, avec une fiabilité du modèle approchant 99,9 % lors de tests rétrospectifs. Des diagrammes de réseaux visuels montrent quelles caractéristiques moléculaires soutiennent le plus fortement un état actif, offrant aux scientifiques une carte interprétable de ce qui motive un comportement prometteur.
Comment cette approche se compare et ce qui vient ensuite
Les auteurs comparent leur méthode à des outils de prédiction en phase précoce populaires tels que pkCSM, SwissADME et ADMETlab, qui estiment l’absorption, la distribution, le métabolisme et l’excrétion d’un médicament. Alors que ces systèmes s’appuient principalement sur des règles prédéfinies ou du machine learning générique, le cadre ASC mesure explicitement la contribution de chaque caractéristique à l’information gagnée sur le comportement des composés semblables à des médicaments et peut simuler des changements de contexte biologique. En parallèle, l’étude note des limites : le jeu de données est relativement petit et fortement déséquilibré, et la méthode n’a jusqu’à présent été appliquée qu’à un seul récepteur. Les auteurs suggèrent que les versions futures pourraient combiner les modèles ASC avec l’apprentissage profond et s’étendre à plusieurs cibles.
Ce que cela signifie pour les médicaments de demain
En termes pratiques, ce travail montre que des modèles riches en information peuvent transformer des données de criblage désordonnées en prédictions claires et testables sur les molécules qui méritent une attention supplémentaire. En nettoyant automatiquement les données, en classant l’importance des caractéristiques et en mettant en lumière des composés rares mais prometteurs, l’approche peut réduire le temps et le coût nécessaires pour atteindre le laboratoire puis, éventuellement, la clinique. Sans remplacer les études animales ou les essais humains, elle agit comme un filtre et un guide puissants, aidant les scientifiques à passer des données brutes à des thérapies potentielles de manière plus efficace et avec une plus grande confiance.
Citation: Saied, H., Alfahad, O., Aljaffer, A.A. et al. From data to discovery: The rise of information-theoretic predictive models in drug development. Sci Rep 16, 12857 (2026). https://doi.org/10.1038/s41598-026-45644-5
Mots-clés: découverte de médicaments, dépistage virtuel, modélisation prédictive, données de bioessais, récepteur peptidique formylé