Clear Sky Science · pt
Explorando os limites dos embeddings pré-treinados no desenho de proteínas guiado por máquina: um estudo de caso sobre a predição da viabilidade de vetores AAV
Por que pequenas alterações em proteínas virais importam
A terapia gênica frequentemente depende de vírus inofensivos, como o vírus adeno-associado (AAV), para transportar genes terapêuticos às nossas células. Tornar esses “veículos de entrega” mais seguros e eficazes costuma exigir ajustar apenas alguns blocos construtores em uma concha proteica muito longa. Este estudo faz uma pergunta aparentemente simples: quando as mudanças importantes são tão pequenas e locais, as poderosas ferramentas de inteligência artificial atuais conseguem enxergá-las bem o suficiente para orientar melhores projetos?
Como os computadores “leem” frases de proteínas
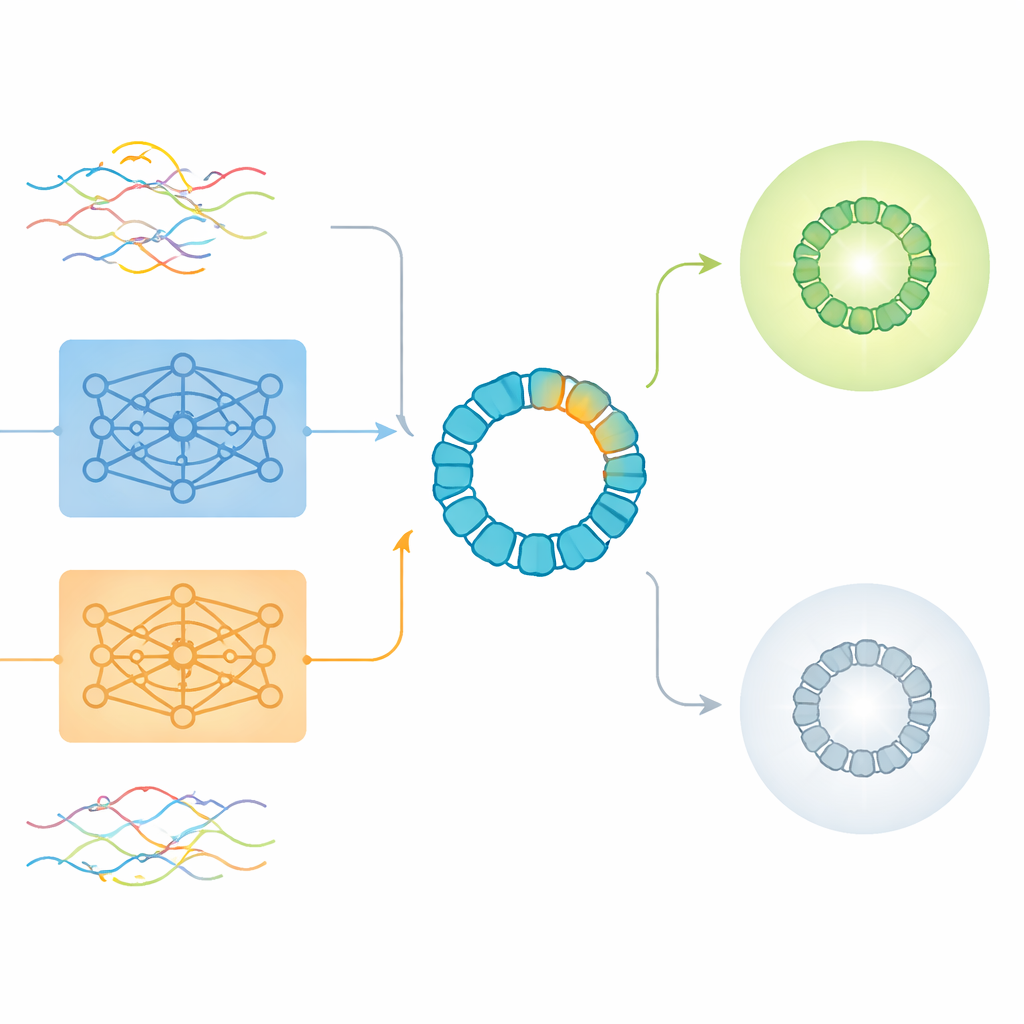
O desenho moderno de proteínas usa frequentemente modelos de deep learning que tratam sequências de aminoácidos de forma análoga a frases em uma linguagem. Ferramentas como ProtBERT e ESM2 aprendem a converter cada proteína em um conjunto de números, chamado embedding, que resume padrões observados em milhões de proteínas naturais. Esses embeddings pré-treinados são atraentes porque capturam informação rica sobre estrutura e função sem exigir novos experimentos. Mas eles foram projetados sobretudo para compreender proteínas inteiras, não as mutações raras porém cruciais que bioengenheiros introduzem em um pequeno trecho.
Testando IA em um cavalo de batalha da terapia gênica
Os autores usaram o AAV2, um vetor de terapia gênica amplamente estudado, como um caso de teste exigente. A casca externa do AAV2, ou capsídeo, é uma proteína longa de 735 aminoácidos, porém os engenheiros normalmente alteram apenas um trecho curto de cerca de 20 a 50 posições para mudar o comportamento do vírus no organismo. A equipe analisou mais de 293.000 variantes medidas experimentalmente cujas mutações estavam confinadas a uma janela de 28 aminoácidos. Cada variante foi rotulada como produzindo partículas virais viáveis ou não. Esse grande conjunto de dados cuidadosamente anotado permitiu aos pesquisadores investigar como diferentes maneiras de codificar sequências — codificações tradicionais one-hot e várias versões de embeddings de ProtBERT e ESM2 — se comportam quando o sinal biológico vem de uma região muito pequena.
O que os embeddings crus deixam passar e onde eles ainda ajudam
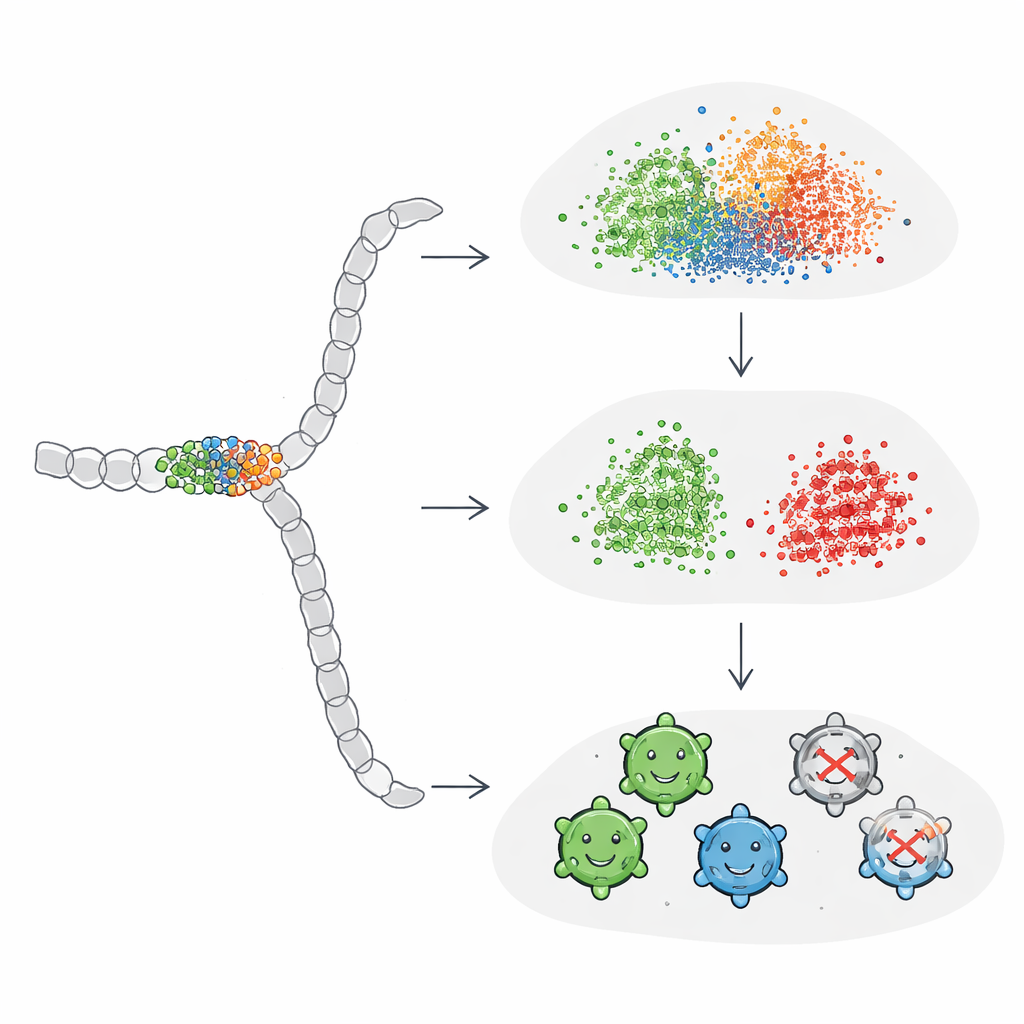
O estudo examinou primeiro análises não supervisionadas, nas quais algoritmos simplesmente agrupam sequências por similaridade sem serem informados sobre quais funcionam. Nessa abordagem, os embeddings globais de sequência gerados pelos modelos de linguagem produziram agrupamentos razoáveis de acordo com como as sequências foram projetadas, mas não separaram claramente variantes viáveis de não viáveis. Codificações one-hot tradicionais tenderam a agrupar proteínas por comprimento, uma propriedade que se mostrou apenas fracamente relacionada à viabilidade. Quando os pesquisadores passaram a tarefas supervisionadas — treinando explicitamente modelos para prever viabilidade — eles descobriram que embeddings ao nível do residu, que agregam informação através de todos os aminoácidos, geralmente superavam os embeddings globais de sequência. Surpreendentemente, entretanto, uma versão comprimida da simples codificação one-hot superou ligeiramente os embeddings pré-treinados em precisão geral, especialmente quando usada com redes neurais.
Por que padrões de mutação são difíceis de enxergar
Para entender esses resultados mistos, os autores examinaram quais variantes todos os modelos concordavam e quais os confundiam consistentemente. Sequências fáceis de classificar eram quase sempre viáveis e mostravam uma clara zona de “não alteração”: variantes bem-sucedidas tendiam a evitar mutações em um segmento estrutural enterrado entre certas posições, ou a limitá-las a substituições sutis. Casos difíceis, por outro lado, pareciam superficialmente semelhantes em onde e quantas mutações carregavam, mas revelavam-se não viáveis. A equipe então construiu exemplos sintéticos espalhando ou concentrando mutações ao longo da proteína. Eles descobriram que embeddings padrão só começaram a separar grupos de forma limpa quando centenas de posições eram alteradas — muito mais do que é prático ou típico em campanhas reais de bioengenharia. Isso indica que embeddings de propósito geral para proteínas são relativamente insensíveis às mutações esparsas ou altamente localizadas que frequentemente fazem ou quebram proteínas projetadas.
Ajustando a IA para focar no que importa
Os pesquisadores então exploraram se poderiam “ensinar” ProtBERT e ESM2 a prestar mais atenção à pequena janela mutacional fazendo fine-tuning dos modelos diretamente com os rótulos de viabilidade do AAV. Eles adicionaram uma camada de classificação simples a cada modelo e treinaram o sistema de ponta a ponta. Após o fine-tuning, o desempenho melhorou a ponto de igualar ou superar os melhores modelos baseados em one-hot, e os embeddings resultantes finalmente mostraram uma separação clara entre sequências viáveis e não viáveis em visualizações. Curiosamente, os embeddings globais de sequência foram os que mais se beneficiaram desse processo: uma vez guiados por feedback específico da tarefa, eles aprenderam a amplificar a influência das posições cruciais em vez de deixá-las ser ofuscadas pelo resto da sequência.
O que isso significa para o desenho futuro de proteínas
Para leitores interessados em como a IA moldará a próxima geração de terapias gênicas e enzimas, a mensagem é matizada, mas otimista. Modelos de linguagem de proteínas prontos para uso, por mais poderosos que sejam, podem negligenciar as mudanças em alta resolução que muitas vezes determinam se uma proteína projetada funciona. Codificações simples e redução de dimensionalidade ainda se mostram competitivas nesses cenários. Contudo, ao ajustar esses modelos com dados experimentais de alta qualidade — mesmo quando as mutações são poucas e fortemente agrupadas — os pesquisadores podem reorientá-los para as partes da sequência que mais importam. Na prática, este trabalho sugere que combinar grandes modelos pré-treinados com retreinamento específico da tarefa oferece um caminho robusto para um projeto mais confiável e guiado por máquina de vetores virais e outras proteínas projetadas.
Citação: Rodrigues, A.F., Ferraz, L., Balbi, L. et al. Exploring the limits of pre-trained embeddings in machine-guided protein design: a case study on predicting AAV vector viability. Sci Rep 16, 10974 (2026). https://doi.org/10.1038/s41598-026-45458-5
Palavras-chave: modelos de linguagem de proteínas, projeto de capsídeo AAV, vetores de terapia gênica, embeddings de proteínas, engenharia de proteínas guiada por máquina