Clear Sky Science · it
Esplorare i limiti degli embedding pre-addestrati nella progettazione proteica guidata da macchine: uno studio di caso sulla predizione della vitalità dei vettori AAV
Perché anche piccole modifiche nelle proteine virali contano
La terapia genica spesso si affida a virus innocui, come l’adeno-associated virus (AAV), per trasportare geni terapeutici nelle nostre cellule. Rendere questi “veicoli” virali più sicuri ed efficaci richiede di solito di modificare solo pochi mattoni in un inviluppo proteico molto lungo. Questo studio pone una domanda apparentemente semplice: quando i cambiamenti importanti sono così minuti e locali, gli strumenti di intelligenza artificiale di oggi riescono davvero a individuarli abbastanza bene da guidare progettazioni migliori?
Come i computer “leggono” le frasi proteiche
La progettazione moderna delle proteine utilizza spesso modelli di deep learning che trattano le sequenze di amminoacidi un po’ come frasi in una lingua. Strumenti come ProtBERT ed ESM2 imparano a convertire ogni proteina in un insieme di numeri, chiamato embedding, che riassume i pattern osservati in milioni di proteine naturali. Questi embedding pre-addestrati sono attraenti perché catturano informazioni ricche su struttura e funzione senza richiedere nuovi esperimenti. Tuttavia, sono stati costruiti principalmente per comprendere proteine intere, non le mutazioni rare ma cruciali che gli ingegneri biologici introducono in una piccola porzione.
Testare l’IA su un cavallo di battaglia della terapia genica
Gli autori hanno utilizzato AAV2, un vettore per terapia genica ampiamente studiato, come caso di prova rigoroso. Il guscio esterno di AAV2, o capside, è una lunga proteina di 735 amminoacidi, eppure gli ingegneri di solito modificano solo un tratto breve di circa 20–50 posizioni per cambiare il comportamento del virus nell’organismo. Il team ha analizzato più di 293.000 varianti misurate sperimentalmente le cui mutazioni erano confinate in una finestra di 28 amminoacidi. Ogni variante è stata etichettata come in grado di produrre particelle virali vitali o incapace di farlo. Questo ampio dataset, accuratamente annotato, ha permesso ai ricercatori di indagare come diversi metodi di codifica delle sequenze—le tradizionali codifiche one-hot e varie tipologie di embedding di ProtBERT ed ESM2—si comportino quando il segnale biologico proviene da una regione molto piccola.
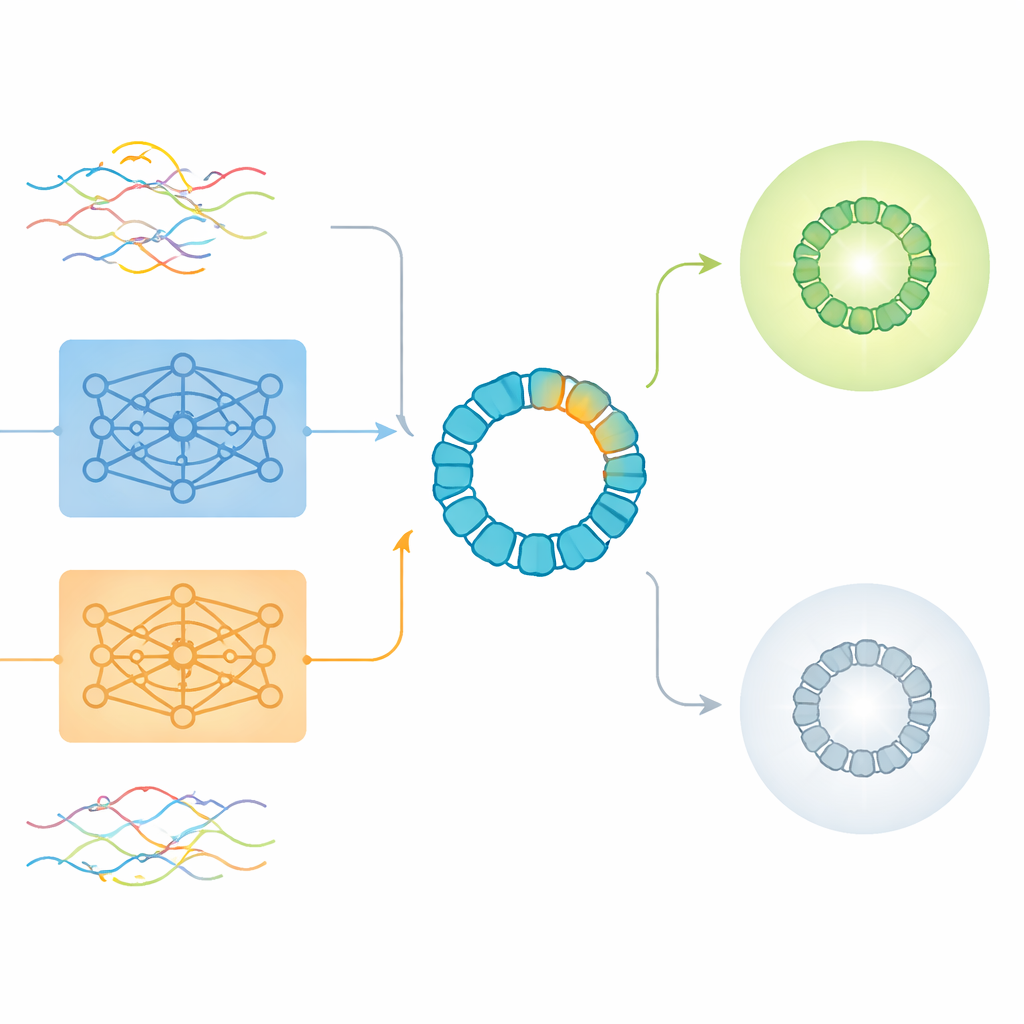
Cosa gli embedding grezzi non colgono e dove sono ancora utili
Lo studio ha prima esaminato analisi non supervisionate, in cui gli algoritmi raggruppano semplicemente le sequenze per similarità senza sapere quali funzionano. Qui, gli embedding globali a livello di sequenza prodotti dai modelli linguistici hanno creato raggruppamenti ragionevoli in base a come le sequenze erano state progettate, ma non hanno separato in modo netto le varianti vitali da quelle non vitali. Le codifiche one-hot tradizionali tendevano invece a raggruppare le proteine per lunghezza, una proprietà risultata solo debolmente correlata alla vitalità. Quando i ricercatori sono passati a compiti supervisionati—addestrando esplicitamente modelli a predire la vitalità—hanno scoperto che gli embedding a livello di singolo amminoacido, che mediavano l’informazione su tutti i residui, generalmente sovraperformavano gli embedding globali della sequenza. Sorprendentemente, però, una versione compressa della semplice codifica one-hot ha superato di poco gli embedding pre-addestrati in termini di accuratezza complessiva, specialmente se usata con reti neurali.
Perché i pattern di mutazione sono difficili da vedere
Per comprendere questi risultati eterogenei, gli autori hanno esaminato quali varianti tutti i modelli concordavano nel classificare e quali invece li confondevano sistematicamente. Le sequenze facili da classificare erano quasi sempre vitali e mostravano una chiara zona “vietata”: le varianti di successo tendevano ad evitare mutazioni in un segmento strutturale sepolto tra determinate posizioni, o le limitavano a sostituzioni sottili. I casi difficili, al contrario, sembravano superficialmente simili per luogo e numero di mutazioni ma si rivelavano non vitali. Il team ha quindi costruito esempi sintetici in cui diffondevano o concentravano le mutazioni lungo l’intera proteina. Hanno scoperto che gli embedding standard cominciavano a separare i gruppi in modo netto solo quando centinaia di posizioni venivano mutate—molto più di quanto sia pratico o tipico nelle campagne di ingegneria biologica reali. Ciò indica che gli embedding proteici di uso generale sono relativamente insensibili alle mutazioni sparse o fortemente localizzate che spesso determinano il successo o il fallimento delle proteine ingegnerizzate.
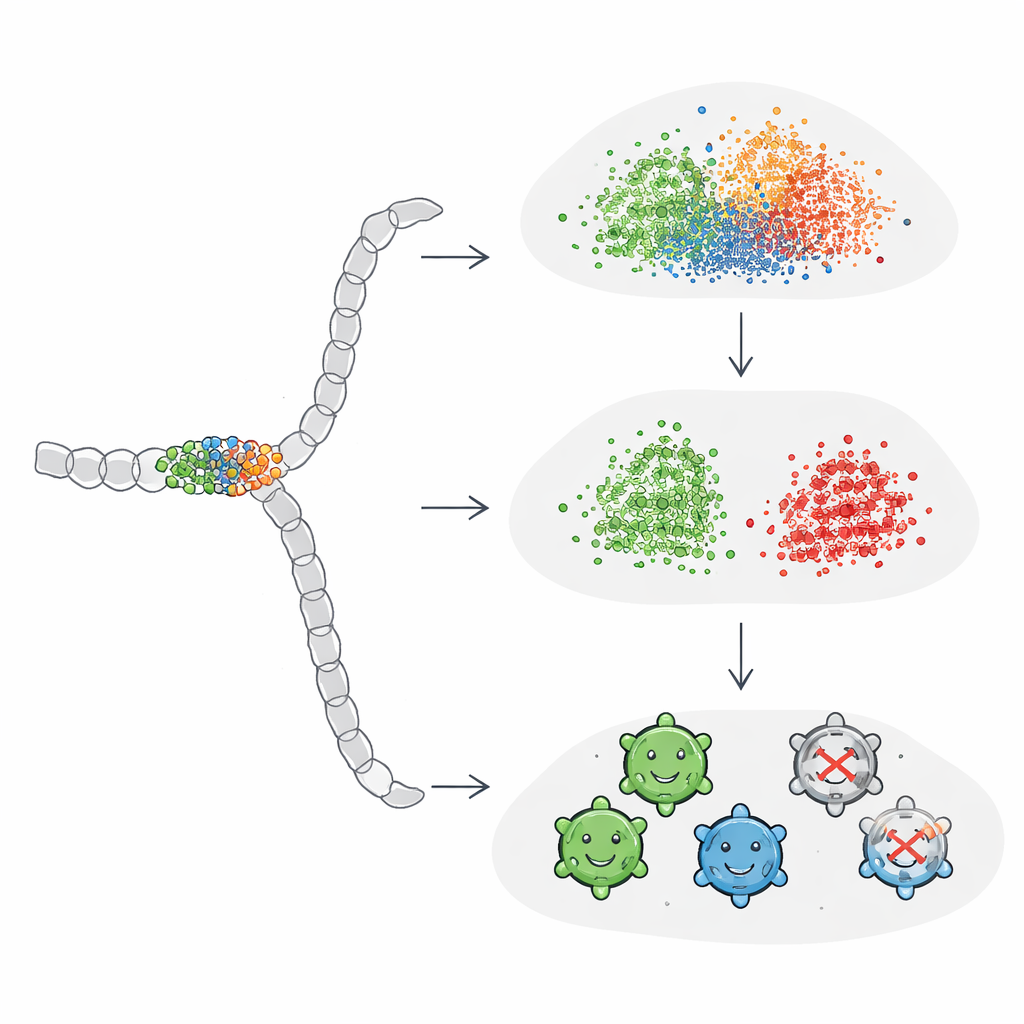
Accordare l’IA per focalizzarsi su ciò che conta
I ricercatori hanno poi esplorato se fosse possibile “insegnare” a ProtBERT ed ESM2 a prestare maggiore attenzione alla piccola finestra mutazionale effettuando un fine-tuning diretto sui label di vitalità AAV. Hanno aggiunto a ciascun modello un semplice strato di classificazione e addestrato l’intero sistema end-to-end. Dopo il fine-tuning, le prestazioni sono migliorate fino a eguagliare o superare i migliori modelli basati su one-hot, e gli embedding risultanti hanno finalmente mostrato una chiara separazione tra sequenze vitali e non vitali nelle visualizzazioni. Interessante notare che gli embedding globali della sequenza hanno beneficiato maggiormente di questo processo: una volta guidati da feedback specifici al compito, hanno imparato ad amplificare l’influenza delle posizioni cruciali invece di lasciarle affogare nel resto della sequenza.
Cosa significa per la progettazione proteica futura
Per i lettori interessati a come l’IA plasmerà la prossima generazione di terapie geniche ed enzimi, il messaggio è sfumato ma promettente. I modelli linguistici proteici pronti all’uso, potenti com’erano, possono trascurare i cambiamenti fini che spesso determinano se una proteina progettata funziona. Codifiche semplici e riduzione dimensionale mantengono ancora la loro efficacia in tali contesti. Eppure, eseguendo il fine-tuning di questi modelli su dati sperimentali di alta qualità—anche quando le mutazioni sono poche e strettamente raggruppate—i ricercatori possono ri-orientarli sulle parti della sequenza che contano di più. In termini pratici, questo lavoro suggerisce che combinare grandi modelli pre-addestrati con un riaddestramento specifico per il compito offre una strada solida verso una progettazione assistita da macchina più affidabile di vettori virali e altre proteine ingegnerizzate.
Citazione: Rodrigues, A.F., Ferraz, L., Balbi, L. et al. Exploring the limits of pre-trained embeddings in machine-guided protein design: a case study on predicting AAV vector viability. Sci Rep 16, 10974 (2026). https://doi.org/10.1038/s41598-026-45458-5
Parole chiave: modelli linguistici proteici, progettazione del capside AAV, vettori per terapia genica, embedding proteici, ingegneria proteica guidata da macchine