Clear Sky Science · es
Explorando los límites de los embeddings preentrenados en el diseño de proteínas guiado por máquinas: un estudio de caso sobre la predicción de la viabilidad de vectores AAV
Por qué importan los cambios minúsculos en las proteínas virales
La terapia génica a menudo depende de virus inocuos, como el virus adenoasociado (AAV), para transportar genes terapéuticos a nuestras células. Hacer que estos “vehículos de entrega” virales sean más seguros y eficaces suele requerir ajustar solo unos pocos bloques constructivos en una cáscara proteica muy larga. Este estudio plantea una pregunta aparentemente simple: cuando los cambios importantes son tan pequeños y localizados, ¿pueden las potentes herramientas de inteligencia artificial actuales verlos con suficiente claridad como para orientar mejores diseños?
Cómo las computadoras leen las “oraciones” de proteínas
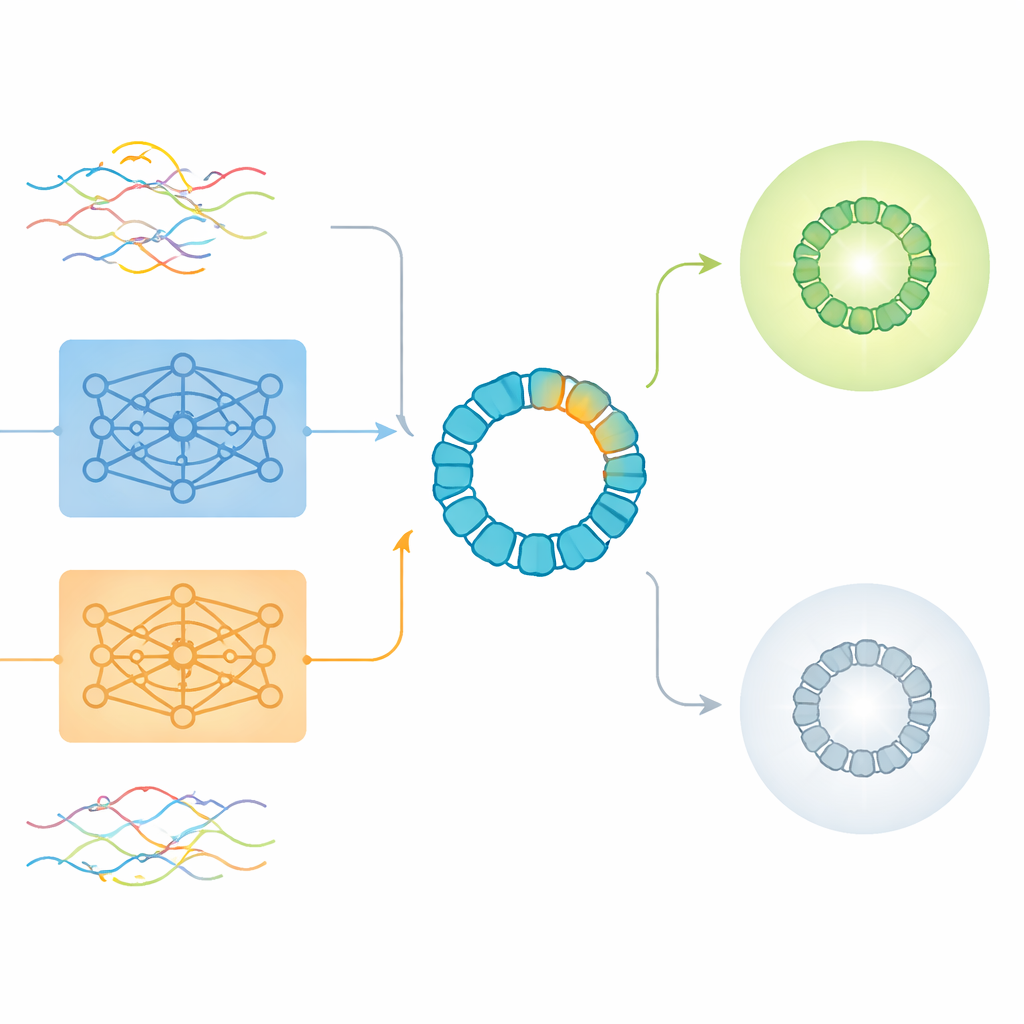
El diseño moderno de proteínas utiliza con frecuencia modelos de aprendizaje profundo que tratan las secuencias de aminoácidos un poco como oraciones en un idioma. Herramientas como ProtBERT y ESM2 aprenden a convertir cada proteína en un conjunto de números, llamado embedding, que resume patrones observados en millones de proteínas naturales. Estos embeddings preentrenados resultan atractivos porque capturan información rica sobre estructura y función sin requerir nuevos experimentos. Pero se construyeron mayoritariamente para comprender proteínas completas, no las mutaciones raras pero cruciales que los bioingenieros introducen en un parche pequeño.
Ponerse a prueba con un caballo de batalla de la terapia génica
Los autores utilizaron AAV2, un vector de terapia génica ampliamente estudiado, como caso de prueba exigente. La cáscara externa de AAV2, o cápside, es una proteína larga de 735 aminoácidos, sin embargo los ingenieros suelen alterar solo un tramo corto de unas 20 a 50 posiciones para cambiar cómo se comporta el virus en el organismo. El equipo analizó más de 293.000 variantes medidas experimentalmente cuyas mutaciones se limitaron a una ventana de 28 aminoácidos. Cada variante se etiquetó como capaz de producir partículas virales viables o como incapaz de hacerlo. Este gran conjunto de datos, cuidadosamente anotado, permitió a los investigadores sondear cómo distintas formas de codificar secuencias —codificaciones one-hot tradicionales y varias versiones de embeddings de ProtBERT y ESM2— rinden cuando la señal biológica proviene de una región muy pequeña.
Lo que los embeddings crudos no captan y dónde siguen siendo útiles
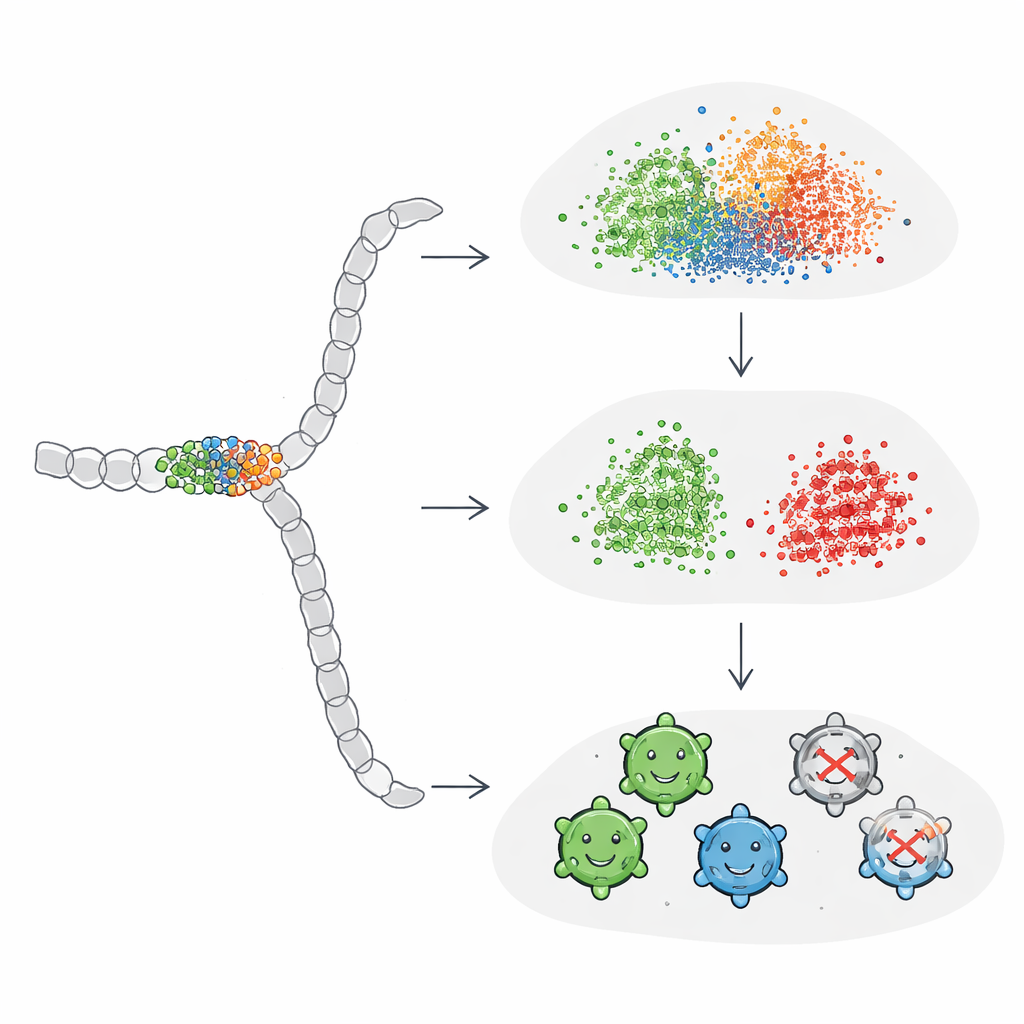
El estudio examinó primero análisis no supervisados, en los que los algoritmos simplemente agrupan secuencias por similitud sin conocer cuáles funcionan. Aquí, los embeddings globales a nivel de secuencia de los modelos de lenguaje produjeron agrupamientos razonables según cómo se habían diseñado las secuencias, pero no separaron claramente las variantes viables de las no viables. Las codificaciones one-hot tradicionales tendieron en cambio a agrupar proteínas por longitud, una propiedad que resultó estar solo débilmente relacionada con la viabilidad. Cuando los investigadores pasaron a tareas supervisadas —entrenando explícitamente modelos para predecir la viabilidad— encontraron que los embeddings a nivel de aminoácido, que agregan información a través de todos los residuos, en general superaron a los embeddings globales de secuencia. Sorprendentemente, sin embargo, una versión comprimida de la simple codificación one-hot superó ligeramente a los embeddings preentrenados en precisión global, especialmente cuando se usó con redes neuronales.
Por qué los patrones de mutación son difíciles de ver
Para entender estos resultados mixtos, los autores examinaron en qué variantes todos los modelos coincidían y cuáles los confundían sistemáticamente. Las secuencias fáciles de clasificar eran casi siempre viables y mostraban una clara zona de “no tocar”: las variantes exitosas tendían a evitar mutaciones en un segmento estructural enterrado entre ciertas posiciones, o las limitaban a sustituciones sutiles. Los casos difíciles, en contraste, parecían superficialmente similares en dónde y cuántas mutaciones presentaban, pero resultaban no viables. El equipo construyó entonces ejemplos sintéticos en los que dispersaron o concentraron mutaciones a lo largo de toda la proteína. Encontraron que los embeddings estándar solo empezaban a separar los grupos de forma nítida cuando se cambiaban cientos de posiciones —mucho más de lo que es práctico o típico en campañas reales de bioingeniería. Esto indica que los embeddings de propósito general son relativamente insensibles a las mutaciones escasas o muy localizadas que a menudo hacen que las proteínas diseñadas funcionen o no.
Ajustar la IA para que se concentre en lo que importa
Los investigadores exploraron a continuación si podían “enseñar” a ProtBERT y ESM2 a prestar más atención a la pequeña ventana mutacional ajustando finamente los modelos directamente con las etiquetas de viabilidad de AAV. Añadieron una capa de clasificación simple a cada modelo y entrenaron todo el sistema de extremo a extremo. Tras el ajuste fino, el rendimiento mejoró hasta igualar o superar a los mejores modelos basados en one-hot, y los embeddings resultantes finalmente mostraron una separación clara entre secuencias viables y no viables en los gráficos de visualización. Curiosamente, los embeddings globales de secuencia fueron los que más se beneficiaron de este proceso: una vez guiados por retroalimentación específica de la tarea, aprendieron a amplificar la influencia de las posiciones cruciales en lugar de dejar que quedaran ahogadas por el resto de la secuencia.
Qué implica esto para el futuro del diseño de proteínas
Para quienes se interesan en cómo la IA moldeará la próxima generación de terapias génicas y enzimas, el mensaje es matizado pero esperanzador. Los modelos de lenguaje de proteínas listos para usar, por potentes que sean, pueden pasar por alto los cambios de grano fino que a menudo determinan si una proteína diseñada funciona. Las codificaciones simples y la reducción de dimensionalidad aún mantienen su valor en esos escenarios. Sin embargo, afinando estos modelos con datos experimentales de alta calidad —incluso cuando las mutaciones son pocas y están muy agrupadas— los investigadores pueden reenfocarlos en las partes de la secuencia que más importan. En términos prácticos, este trabajo sugiere que combinar grandes modelos preentrenados con reentrenamiento específico de la tarea ofrece una vía sólida hacia un diseño más fiable y guiado por máquinas de vectores virales y otras proteínas diseñadas.
Cita: Rodrigues, A.F., Ferraz, L., Balbi, L. et al. Exploring the limits of pre-trained embeddings in machine-guided protein design: a case study on predicting AAV vector viability. Sci Rep 16, 10974 (2026). https://doi.org/10.1038/s41598-026-45458-5
Palabras clave: modelos de lenguaje de proteínas, diseño de cápsides AAV, vectores de terapia génica, embeddings de proteínas, ingeniería de proteínas guiada por máquinas