Clear Sky Science · en
Exploring the limits of pre-trained embeddings in machine-guided protein design: a case study on predicting AAV vector viability
Why tiny changes in viral proteins matter
Gene therapy often relies on harmless viruses, such as adeno-associated virus (AAV), to carry therapeutic genes into our cells. Making these viral “delivery vehicles” safer and more effective usually requires tweaking only a few building blocks in a very long protein shell. This study asks a deceptively simple question: when the important changes are so tiny and local, can today’s powerful artificial intelligence tools actually see them well enough to guide better designs?
How computers read protein “sentences”
Modern protein design frequently uses deep learning models that treat amino acid sequences a bit like sentences in a language. Tools such as ProtBERT and ESM2 learn to convert each protein into a bundle of numbers, called an embedding, that summarizes patterns they have seen across millions of natural proteins. These pre-trained embeddings are attractive because they capture rich information about structure and function without requiring new experiments. But they were mostly built to understand whole proteins, not the rare but crucial mutations that bioengineers introduce in just a small patch.
Testing AI on a real gene therapy workhorse
The authors used AAV2, a widely studied gene therapy vector, as a stringent test case. AAV2’s outer shell, or capsid, is a long protein of 735 amino acids, yet engineers usually alter only a short stretch of about 20 to 50 positions to change how the virus behaves in the body. The team analyzed more than 293,000 experimentally measured variants whose mutations were confined to a 28–amino acid window. Each variant was labeled as either producing viable virus particles or failing to do so. This large, carefully annotated dataset allowed the researchers to probe how different ways of encoding sequences—traditional one-hot encodings and several flavors of ProtBERT and ESM2 embeddings—perform when the biological signal comes from a very small region.
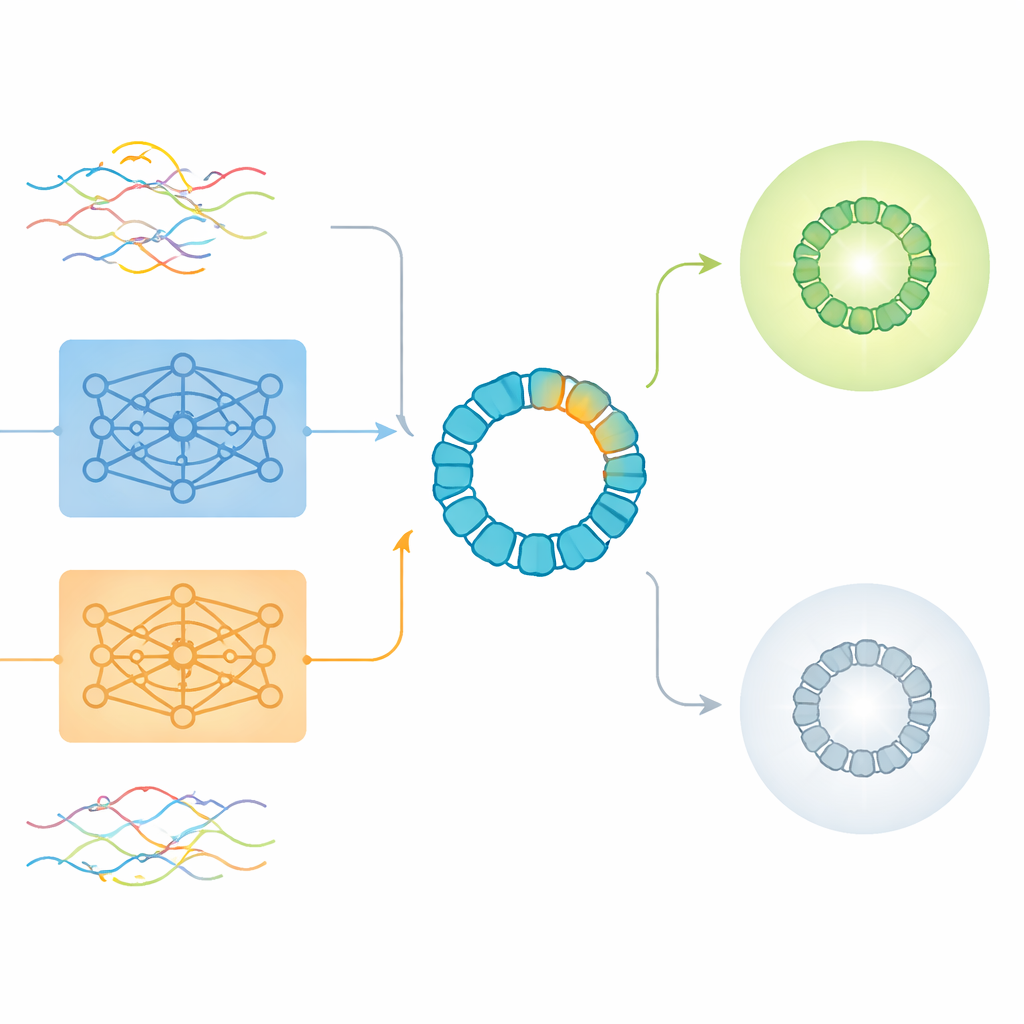
What raw embeddings miss and where they still help
The study first looked at unsupervised analyses, in which algorithms simply group sequences by similarity without being told which ones work. Here, global sequence-level embeddings from the language models produced reasonable groupings according to how the sequences had been designed, but they did not clearly separate viable from non-viable variants. Traditional one-hot encodings tended instead to group proteins by length, a property that turned out to be only weakly related to viability. When the researchers turned to supervised tasks—explicitly training models to predict viability—they found that amino-acid–level embeddings, which average information across all residues, generally outperformed global sequence embeddings. Surprisingly, however, a compressed version of simple one-hot encoding slightly edged out the pre-trained embeddings in overall accuracy, especially when used with neural networks.
Why mutation patterns are hard to see
To understand these mixed results, the authors examined which variants all models agreed on and which consistently confused them. Easy-to-classify sequences were almost always viable and showed a clear “no-go” zone: successful variants tended to avoid mutations in a buried structural segment between certain positions, or limited them to subtle substitutions. Difficult cases, by contrast, looked superficially similar in where and how many mutations they carried but turned out to be non-viable. The team then built synthetic examples where they spread or concentrated mutations along the entire protein. They found that standard embeddings only started to separate groups cleanly when hundreds of positions were changed—far more than is practical or typical in real bioengineering campaigns. This indicates that general-purpose protein embeddings are relatively insensitive to the sparse or highly localized mutations that often make or break engineered proteins.
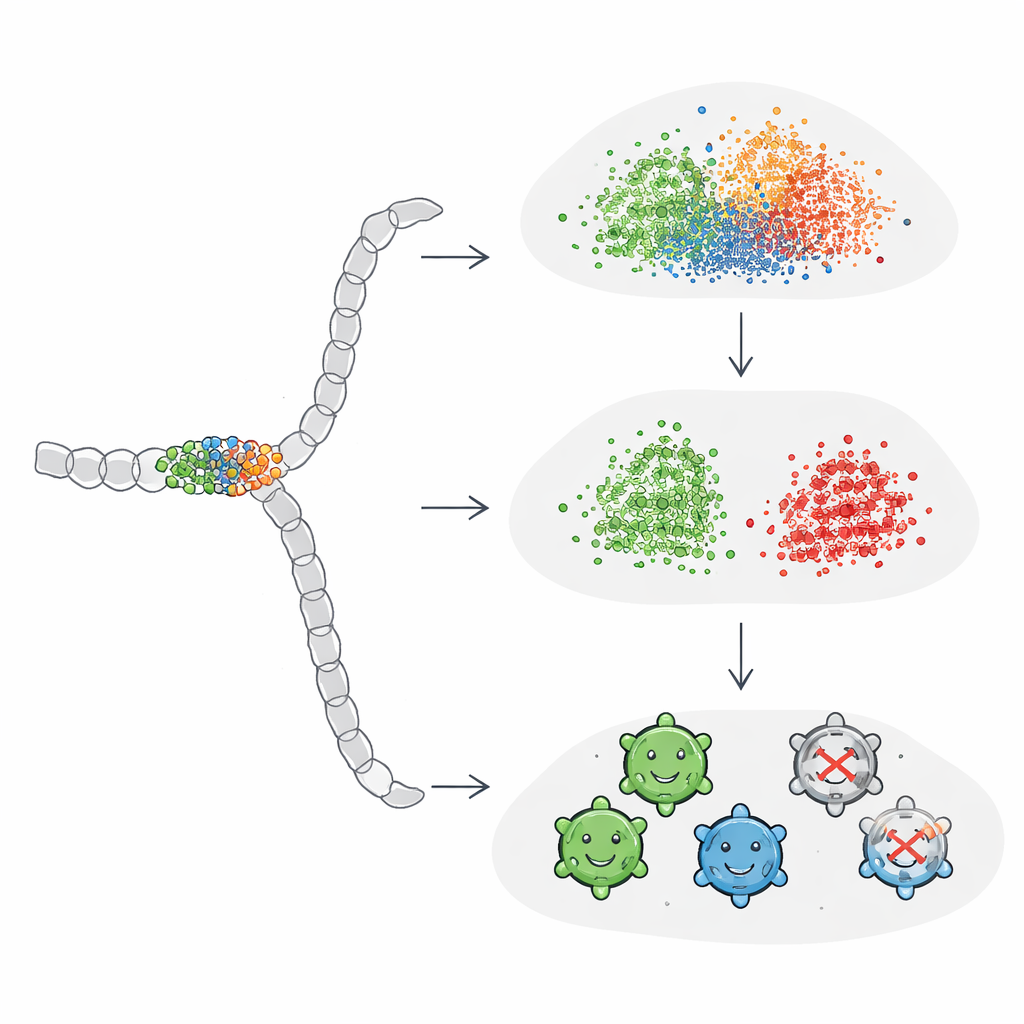
Tuning AI to focus on what matters
The researchers next explored whether they could “teach” ProtBERT and ESM2 to pay closer attention to the small mutational window by fine-tuning the models directly on the AAV viability labels. They attached a simple classification layer to each model and trained the whole system end-to-end. After fine-tuning, performance improved to match or surpass the best one-hot–based models, and the resulting embeddings finally showed a clear separation between viable and non-viable sequences in visualization plots. Interestingly, global sequence embeddings benefited the most from this process: once guided by task-specific feedback, they learned to amplify the influence of the crucial positions instead of letting them be drowned out by the rest of the sequence.
What this means for future protein design
For readers interested in how AI will shape the next generation of gene therapies and enzymes, the message is nuanced but hopeful. Out-of-the-box protein language models, powerful as they are, can overlook the fine-grained changes that often determine whether a designed protein works. Simple encodings and dimensionality reduction still hold their own in such settings. Yet, by fine-tuning these models on high-quality experimental data—even when mutations are few and tightly clustered—researchers can re-focus them on the parts of the sequence that matter most. In practical terms, this work suggests that combining large pre-trained models with task-specific retraining offers a robust path toward more reliable, machine-guided design of viral vectors and other engineered proteins.
Citation: Rodrigues, A.F., Ferraz, L., Balbi, L. et al. Exploring the limits of pre-trained embeddings in machine-guided protein design: a case study on predicting AAV vector viability. Sci Rep 16, 10974 (2026). https://doi.org/10.1038/s41598-026-45458-5
Keywords: protein language models, AAV capsid design, gene therapy vectors, protein embeddings, machine-guided protein engineering