Clear Sky Science · de
Die Grenzen vortrainierter Embeddings beim maschinengestützten Proteindesign erkunden: Eine Fallstudie zur Vorhersage der Lebensfähigkeit von AAV-Vektoren
Warum winzige Veränderungen in viralen Proteinen von Bedeutung sind
Gentherapie nutzt häufig harmlose Viren wie das adeno-assoziierte Virus (AAV), um therapeutische Gene in unsere Zellen zu transportieren. Um diese viralen „Transportsysteme“ sicherer und wirksamer zu machen, genügt es oft, nur wenige Bausteine in einer sehr langen Proteinhülle zu verändern. Diese Studie stellt eine auf den ersten Blick einfache Frage: Wenn die wichtigen Änderungen so winzig und lokal sind, können heutige leistungsfähige KI‑Werkzeuge sie tatsächlich gut genug erkennen, um bessere Designs zu ermöglichen?
Wie Computer Protein-„Sätze“ lesen
Moderne Proteingestaltung nutzt häufig Deep‑Learning‑Modelle, die Aminosäuresequenzen etwas wie Sätze in einer Sprache behandeln. Werkzeuge wie ProtBERT und ESM2 lernen, jedes Protein in ein Bündel Zahlen zu übersetzen — ein sogenanntes Embedding — das Muster zusammenfasst, die sie in Millionen natürlicher Proteine gesehen haben. Diese vortrainierten Embeddings sind attraktiv, weil sie reichhaltige Informationen über Struktur und Funktion erfassen, ohne neue Experimente zu erfordern. Sie wurden jedoch überwiegend dafür entwickelt, ganze Proteine zu verstehen, nicht die seltenen, aber entscheidenden Mutationen, die Bioingenieur*innen meist nur in einem kleinen Abschnitt einführen.
KI an einem realen Arbeitspferd der Gentherapie testen
Die Autor*innen verwendeten AAV2, einen in der Gentherapie intensiv untersuchten Vektor, als strengen Testfall. Die Außenhülle (Kapsid) von AAV2 ist ein langes Protein mit 735 Aminosäuren, doch Ingenieur*innen verändern typischerweise nur einen kurzen Abschnitt von etwa 20 bis 50 Positionen, um das Verhalten des Virus im Körper zu ändern. Das Team analysierte mehr als 293.000 experimentell gemessene Varianten, deren Mutationen auf ein 28‑Aminosäuren‑Fenster begrenzt waren. Jede Variante wurde als entweder produzierte lebensfähige Partikel oder als nicht lebensfähig klassifiziert. Dieser große, sorgfältig annotierte Datensatz erlaubte den Forschenden zu prüfen, wie verschiedene Arten der Sequenzkodierung — traditionelle One‑Hot‑Kodierungen und mehrere Varianten von ProtBERT‑ und ESM2‑Embeddings — abschneiden, wenn das biologische Signal aus einem sehr kleinen Bereich stammt.
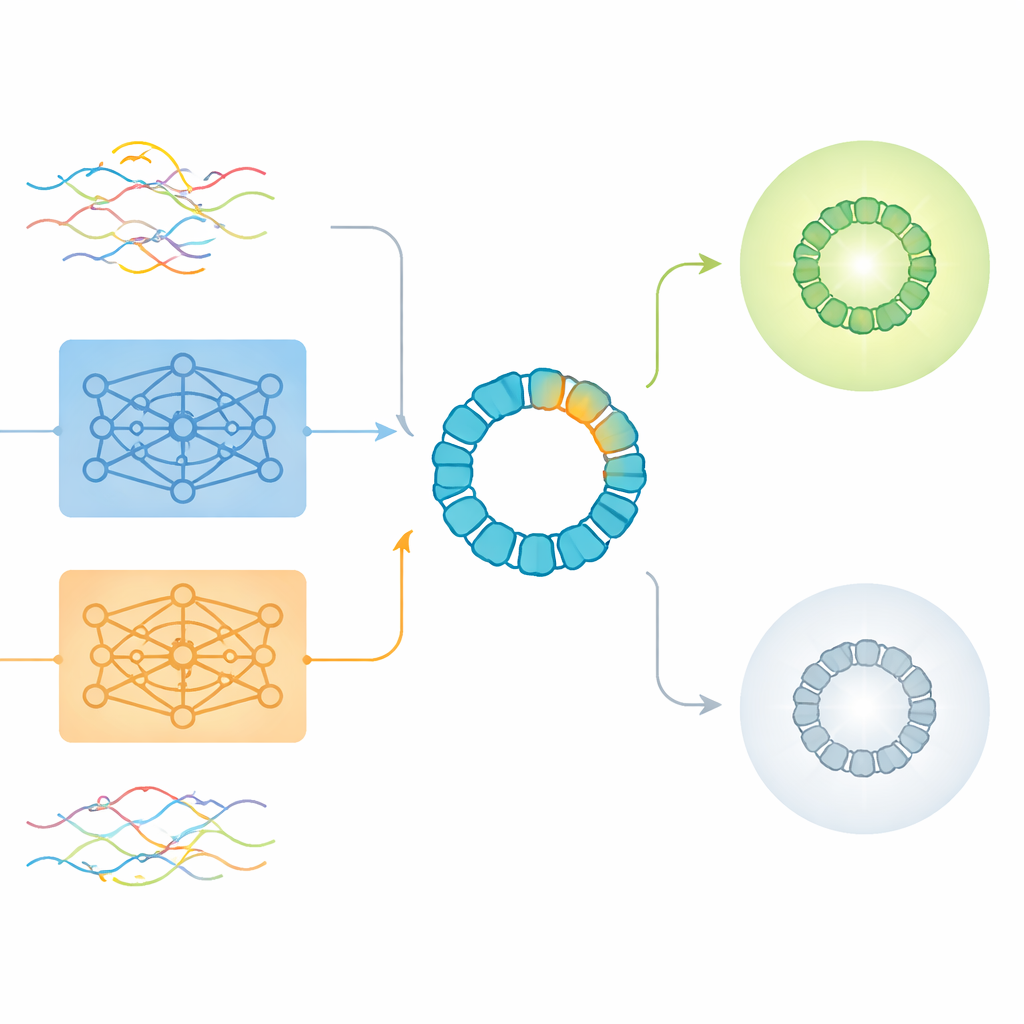
Was rohe Embeddings verpassen und wo sie trotzdem helfen
Die Studie betrachtete zunächst unüberwachte Analysen, bei denen Algorithmen Sequenzen einfach nach Ähnlichkeit gruppieren, ohne zu wissen, welche funktionieren. Hier produzierten globale Sequenz‑Embeddings aus den Sprachmodellen brauchbare Gruppierungen in Abhängigkeit davon, wie die Sequenzen entworfen wurden, trennten jedoch lebensfähige von nicht‑lebensfähigen Varianten nicht deutlich. Traditionelle One‑Hot‑Kodierungen gruppierten eher nach Proteinklänge, einer Eigenschaft, die sich als nur schwach mit Lebensfähigkeit korrelierend erwies. In überwachten Aufgaben — beim expliziten Trainieren von Modellen zur Vorhersage der Lebensfähigkeit — zeigten Aminosäure‑Level‑Embeddings, die Informationen über alle Reste mitteln, im Allgemeinen bessere Leistung als globale Sequenz‑Embeddings. Überraschenderweise schnitt jedoch eine komprimierte Form einfacher One‑Hot‑Kodierung in der Gesamtgenauigkeit leicht besser ab als die vortrainierten Embeddings, insbesondere in Kombination mit neuronalen Netzen.
Warum Mutationsmuster schwer zu erkennen sind
Um diese gemischten Ergebnisse zu erklären, untersuchten die Autor*innen, bei welchen Varianten alle Modelle übereinstimmten und welche sie konsistent verwirrten. Leicht zu klassifizierende Sequenzen waren fast immer lebensfähig und zeigten eine klare „No‑Go“-Zone: Erfolgreiche Varianten vermieden tendenziell Mutationen in einem eingegrabenen Struktursegment zwischen bestimmten Positionen oder beschränkten sie auf subtile Substitutionen. Schwierige Fälle dagegen sahen oberflächlich ähnlich aus hinsichtlich Ort und Anzahl der Mutationen, erwiesen sich aber als nicht lebensfähig. Das Team baute anschließend synthetische Beispiele, in denen sie Mutationen über das gesamte Protein verteilten oder konzentrierten. Sie fanden, dass Standard‑Embeddings Gruppen erst dann sauber zu trennen begannen, wenn Hunderte von Positionen verändert wurden — weit mehr, als in der Praxis oder typischen Bioingenieur‑Projekten vorkommt. Das deutet darauf hin, dass universelle Protein‑Embeddings relativ unempfindlich gegenüber sparsamen oder stark lokalisierten Mutationen sind, die oft über Erfolg oder Misserfolg von konstruierten Proteinen entscheiden.
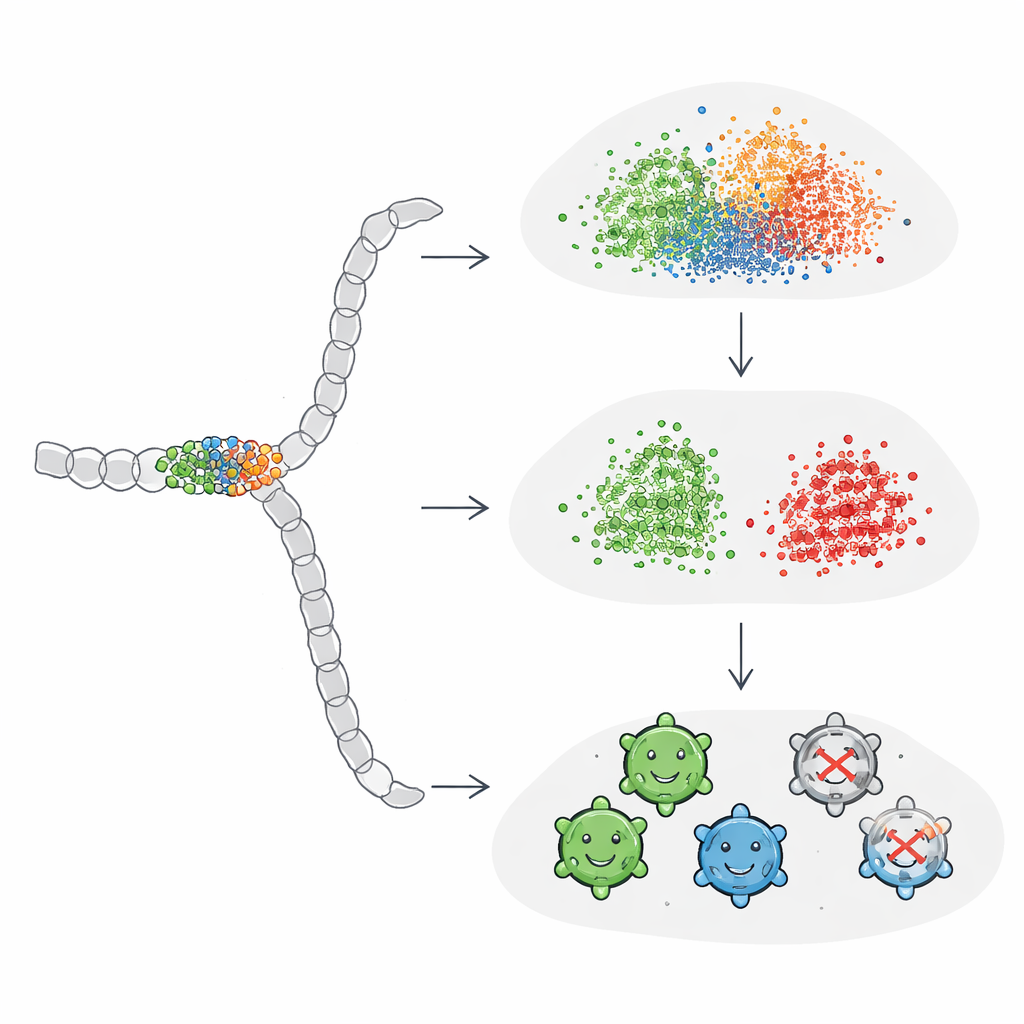
Die KI auf das Wichtige ausrichten
Die Forschenden untersuchten anschließend, ob sie ProtBERT und ESM2 durch Feinabstimmung gezielt auf das kleine Mutationsfenster fokussieren können, indem sie die Modelle direkt mit den AAV‑Lebensfähigkeitslabels weitertrainierten. Sie hängten jeder Modellarchitektur eine einfache Klassifikationsschicht an und trainierten das Gesamtsystem End‑to‑End. Nach der Feinabstimmung verbesserte sich die Leistung, sodass sie mit den besten One‑Hot‑basierten Modellen gleichzog oder diese übertraf, und die resultierenden Embeddings zeigten schließlich in Visualisierungen eine klare Trennung zwischen lebensfähigen und nicht‑lebensfähigen Sequenzen. Interessanterweise profitierten globale Sequenz‑Embeddings am stärksten von diesem Prozess: Durch die aufgabenspezifische Rückkopplung lernten sie, den Einfluss der entscheidenden Positionen zu verstärken, statt ihn vom Rest der Sequenz übertönen zu lassen.
Was das für zukünftiges Protein‑Design bedeutet
Für Leserinnen und Leser, die sich dafür interessieren, wie KI die nächste Generation von Gentherapien und Enzymen prägen wird, ist die Botschaft nuanciert, aber hoffnungsvoll. Out‑of‑the‑box Protein‑Sprachmodelle sind zwar mächtig, können aber die feinen Veränderungen übersehen, die oft entscheiden, ob ein konstruiertes Protein funktioniert. Einfache Kodierungen und Dimensionsreduktion behaupten sich in solchen Szenarien weiterhin. Doch durch Feinabstimmung dieser Modelle mit hochwertiger experimenteller Daten — selbst wenn Mutationen wenige und eng konzentriert sind — können Forschende sie wieder auf die Sequenzbereiche ausrichten, die am wichtigsten sind. Praktisch betrachtet legt diese Arbeit nahe, dass die Kombination großer vortrainierter Modelle mit aufgabenspezifischem Retraining einen robusten Weg zu zuverlässigeren, maschinengestützten Designs von viralen Vektoren und anderen konstruierten Proteinen bietet.
Zitation: Rodrigues, A.F., Ferraz, L., Balbi, L. et al. Exploring the limits of pre-trained embeddings in machine-guided protein design: a case study on predicting AAV vector viability. Sci Rep 16, 10974 (2026). https://doi.org/10.1038/s41598-026-45458-5
Schlüsselwörter: Protein-Sprachmodelle, AAV-Kapsid-Design, Vektoren für Gentherapie, Protein-Embeddings, maschinengestützte Proteintechnik