Clear Sky Science · fr
Explorer les limites des embeddings pré-entraînés dans la conception assistée par machine de protéines : étude de cas sur la prédiction de la viabilité des vecteurs AAV
Pourquoi de minuscules changements dans les protéines virales comptent
La thérapie génique repose souvent sur des virus inoffensifs, comme le virus adéno-associé (AAV), pour transporter des gènes thérapeutiques dans nos cellules. Rendre ces « véhicules de livraison » viraux plus sûrs et plus efficaces nécessite généralement de modifier seulement quelques blocs de construction dans une coque protéique très longue. Cette étude pose une question apparemment simple : lorsque les changements importants sont si infimes et localisés, les outils d’intelligence artificielle actuels peuvent-ils réellement les détecter suffisamment bien pour orienter de meilleurs designs ?
Comment les ordinateurs lisent les « phrases » protéiques
La conception moderne de protéines utilise fréquemment des modèles d’apprentissage profond qui traitent les séquences d’acides aminés un peu comme des phrases dans une langue. Des outils tels que ProtBERT et ESM2 apprennent à convertir chaque protéine en un ensemble de nombres, appelé embedding, qui résume des motifs observés à travers des millions de protéines naturelles. Ces embeddings pré-entraînés sont attractifs car ils capturent des informations riches sur la structure et la fonction sans nécessiter de nouvelles expériences. Mais ils ont été principalement conçus pour comprendre des protéines entières, et non les rares mais cruciales mutations que les bioingénieurs introduisent sur une petite région.
Tester l’IA sur un outil de la thérapie génique
Les auteurs ont utilisé l’AAV2, un vecteur de thérapie génique largement étudié, comme cas-test exigeant. La coque externe de l’AAV2, ou capsid, est une longue protéine de 735 acides aminés, pourtant les ingénieurs modifient généralement seulement un court segment d’environ 20 à 50 positions pour changer le comportement du virus dans l’organisme. L’équipe a analysé plus de 293 000 variantes mesurées expérimentalement dont les mutations étaient confinées à une fenêtre de 28 acides aminés. Chaque variante était étiquetée comme produisant des particules virales viables ou échouant à le faire. Cet ensemble de données large et soigneusement annoté a permis aux chercheurs d’examiner comment différentes façons d’encoder les séquences — encodages one-hot traditionnels et plusieurs variantes d’embeddings ProtBERT et ESM2 — se comportent lorsque le signal biologique provient d’une région très petite.
Ce que les embeddings bruts manquent et où ils aident encore
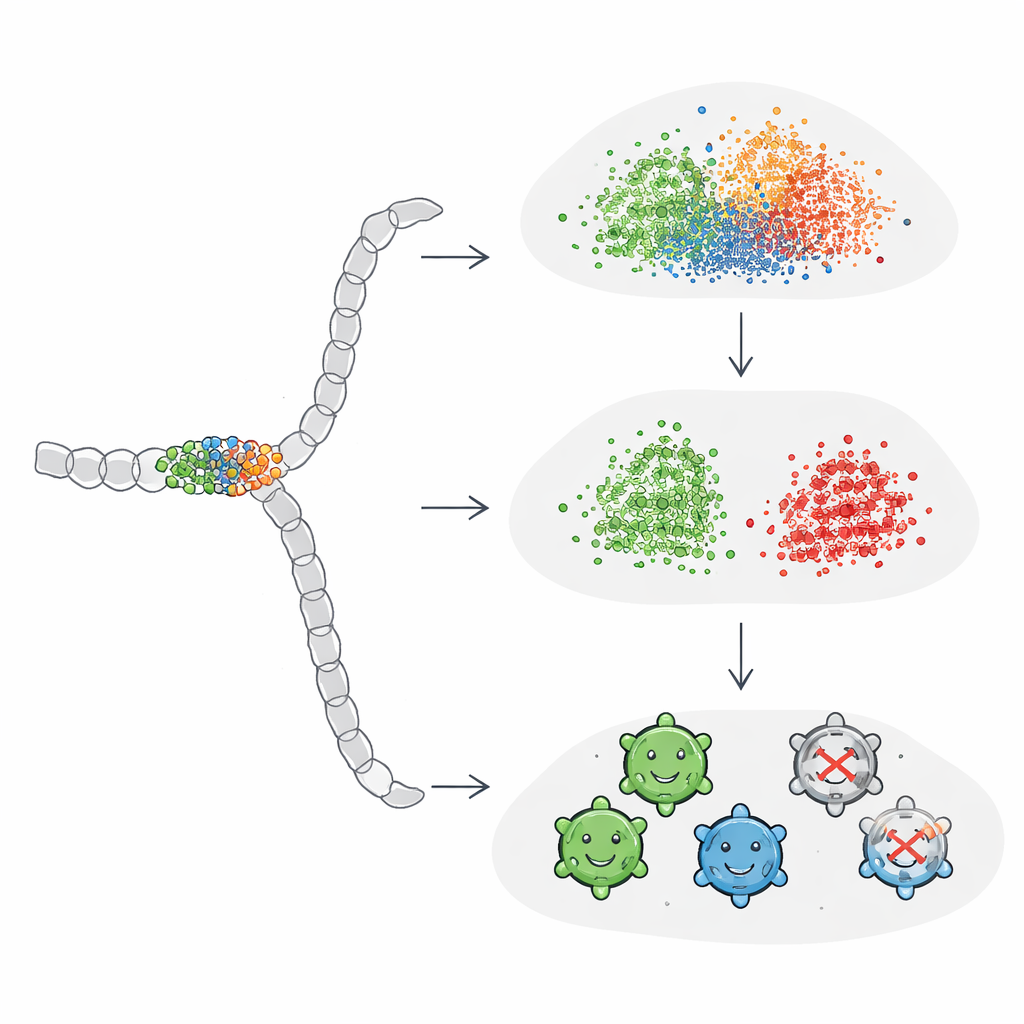
L’étude a d’abord examiné des analyses non supervisées, dans lesquelles les algorithmes regroupent simplement les séquences par similarité sans savoir lesquelles fonctionnent. Ici, les embeddings globaux de niveau séquence issus des modèles de langage ont produit des groupements raisonnables en fonction de la façon dont les séquences avaient été conçues, mais ils ne séparaient pas clairement les variantes viables des non viables. Les encodages one-hot traditionnels avaient plutôt tendance à regrouper les protéines par longueur, une propriété qui s’est avérée faiblement corrélée à la viabilité. Lorsque les chercheurs sont passés à des tâches supervisées — entraînant explicitement des modèles à prédire la viabilité — ils ont trouvé que les embeddings au niveau des acides aminés, qui moyennent l’information sur tous les résidus, surpassaient généralement les embeddings globaux de séquence. De façon surprenante, cependant, une version compressée du simple encodage one-hot devançait légèrement les embeddings pré-entraînés en précision globale, surtout lorsqu’elle était utilisée avec des réseaux neuronaux.
Pourquoi les motifs de mutation sont difficiles à voir
Pour comprendre ces résultats mitigés, les auteurs ont examiné sur quelles variantes tous les modèles étaient d’accord et lesquelles les confondaient systématiquement. Les séquences faciles à classer étaient presque toujours viables et montraient une claire zone « interdite » : les variantes réussies évitaient les mutations dans un segment structurel enfoui entre certaines positions, ou limitaient celles-ci à des substitutions subtiles. Les cas difficiles, en revanche, semblaient superficiellement similaires quant à l’emplacement et au nombre de mutations qu’ils portaient mais s’avéraient non viables. L’équipe a ensuite construit des exemples synthétiques où ils répartissaient ou concentraient les mutations le long de la protéine entière. Ils ont constaté que les embeddings standards ne commençaient à séparer les groupes de façon nette que lorsque des centaines de positions étaient modifiées — bien plus que ce qui est pratique ou typique dans de vraies campagnes de bioingénierie. Cela indique que les embeddings protéiques à usage général sont relativement insensibles aux mutations rares ou fortement localisées qui font souvent la différence pour les protéines conçues.
Régler l’IA pour qu’elle se concentre sur l’essentiel
Les chercheurs ont ensuite exploré s’ils pouvaient « apprendre » à ProtBERT et ESM2 à prêter plus d’attention à la petite fenêtre de mutations en affinant les modèles directement sur les étiquettes de viabilité AAV. Ils ont ajouté une simple couche de classification à chaque modèle et entraîné l’ensemble de bout en bout. Après ce fine-tuning, les performances se sont améliorées pour égaler ou dépasser les meilleurs modèles basés sur one-hot, et les embeddings résultants ont finalement montré une séparation claire entre séquences viables et non viables dans les visualisations. Fait intéressant, les embeddings globaux de séquence ont le plus profité de ce processus : une fois guidés par un retour d’information spécifique à la tâche, ils ont appris à amplifier l’influence des positions cruciales au lieu de les laisser noyées par le reste de la séquence.
Ce que cela implique pour la conception future des protéines
Pour les lecteurs intéressés par la façon dont l’IA influencera la prochaine génération de thérapies géniques et d’enzymes, le message est nuancé mais porteur d’espoir. Les modèles de langage protéique prêts à l’emploi, aussi puissants soient-ils, peuvent négliger les changements fins qui déterminent souvent si une protéine conçue fonctionne. Les encodages simples et la réduction de dimension conservent encore leur pertinence dans de tels contextes. Pourtant, en affinant ces modèles sur des données expérimentales de haute qualité — même lorsque les mutations sont peu nombreuses et fortement regroupées — les chercheurs peuvent les recentrer sur les parties de la séquence qui comptent le plus. En termes pratiques, ce travail suggère que combiner de grands modèles pré-entraînés avec un réentraînement spécifique à la tâche offre une voie robuste vers une conception assistée par machine plus fiable de vecteurs viraux et d’autres protéines conçues.
Citation: Rodrigues, A.F., Ferraz, L., Balbi, L. et al. Exploring the limits of pre-trained embeddings in machine-guided protein design: a case study on predicting AAV vector viability. Sci Rep 16, 10974 (2026). https://doi.org/10.1038/s41598-026-45458-5
Mots-clés: modèles de langage protéique, conception de capsides AAV, vecteurs de thérapie génique, embeddings protéiques, ingénierie des protéines assistée par machine