Clear Sky Science · pt
STHELAR, um conjunto de dados multitecido que conecta transcriptômica espacial e histologia para anotação de tipos celulares
Por que olhar o câncer ao microscópio não é suficiente
Os médicos que tratam câncer ainda dependem muito do que veem ao microscópio: cortes finos de tecido corados em tons de rosa e roxo com hematoxilina e eosina (H&E). Essas imagens revelam formas e padrões, mas não mostram diretamente quais genes estão ativos em cada célula. Um novo recurso chamado STHELAR une esses dois mundos, relacionando a visão familiar das lâminas de tecido com medições “espaciais” de ponta da atividade gênica. Para o leitor, esse trabalho é importante porque abre caminho para ferramentas mais rápidas e mais baratas que, no futuro, podem inferir a composição molecular de tumores a partir de imagens digitais comuns.
Ver o tumor como um bairro lotado
Os tumores não são apenas células cancerosas fora de controle; são bairros lotados repletos de células imunes, vasos sanguíneos, células de suporte e tecido normal. A mistura e a disposição desses habitantes — o microambiente tumoral — podem influenciar como um câncer cresce e como responde ao tratamento. A transcriptômica espacial, uma tecnologia recente, pode mapear quais genes estão ativados em células individuais mantendo sua posição precisa no tecido. No entanto, esses experimentos são caros e tecnicamente exigentes, por isso ainda não fazem parte do cuidado rotineiro. Em contraste, digitalizações de alta resolução de lâminas H&E são hoje comuns, baratas de armazenar e já usadas em todo o mundo. A ideia central por trás do STHELAR é usar um conjunto limitado de experimentos de transcriptômica espacial como um “professor” para milhões de células vistas em imagens H&E padrão.
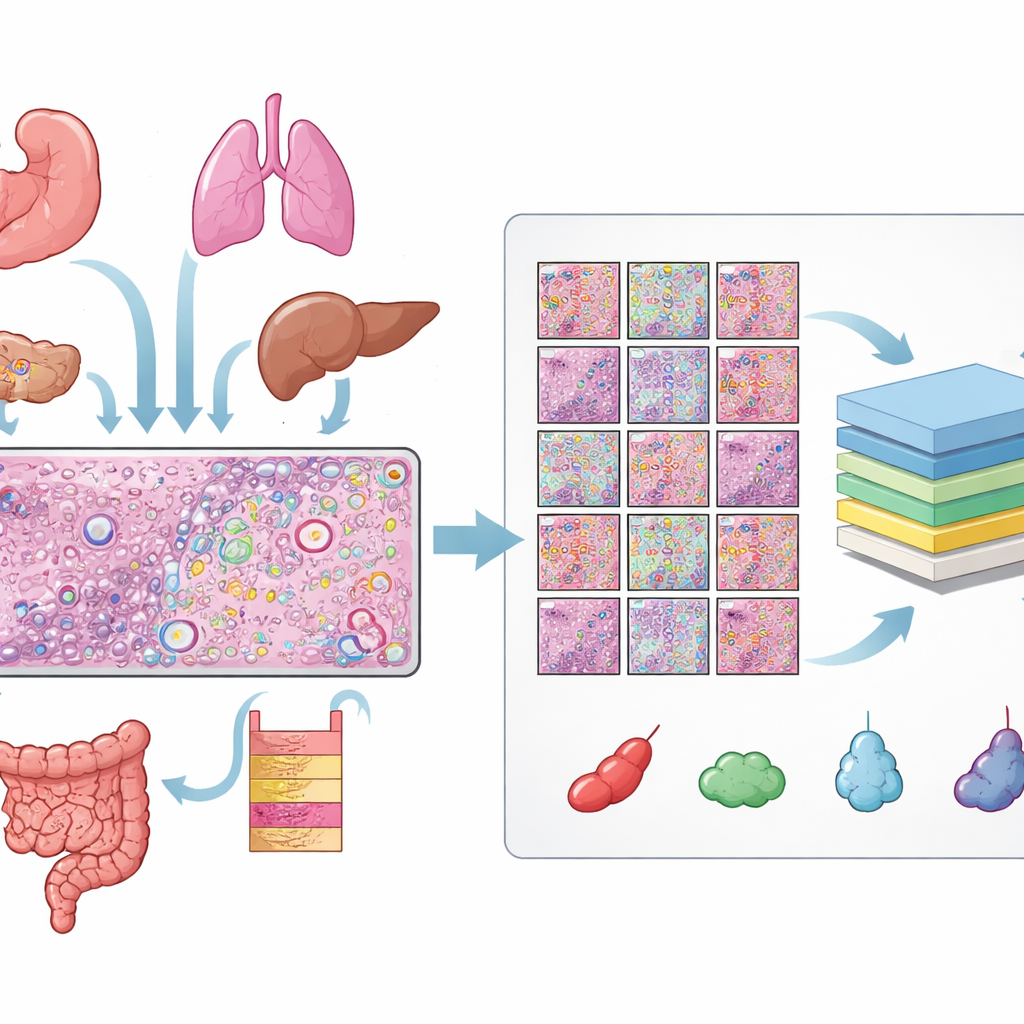
Construindo uma enorme biblioteca de células rotuladas
Os autores reuniram dados de 31 seções de tecido mensuradas com a plataforma de transcriptômica espacial Xenium da 10x Genomics, cobrindo 16 tipos de tecido humano e 22 amostras cancerosas e 9 não cancerosas. Para cada seção, eles dispunham de três visões compatíveis: uma lâmina H&E, uma imagem fluorescente mostrando os núcleos celulares e um mapa de moléculas individuais de RNA. Alinhar essas vistas exigiu verificações de qualidade cuidadosas e, para muitas lâminas, ajustes manuais finos para que cada núcleo na imagem fluorescente correspondesse à estrutura equivalente na lâmina H&E. A partir dessas imagens alinhadas, obtiveram mais de 11 milhões de células distintas e mais de meio milhão de pequenos blocos (patches) H&E, cada um com contornos precisos de cada núcleo.
Ensinando ao computador o que é cada tipo celular
Saber onde cada célula está não basta; o passo crucial é decidir que tipo de célula é. Para isso, a equipe combinou transcriptômica espacial com grandes catálogos existentes de perfis de RNA de célula única. Usando um método chamado Tangram, eles primeiro transferiram identidades celulares prováveis desses atlas de referência para os dados espaciais. Em seguida, aperfeiçoaram esses rótulos preliminares agrupando células em clusters com base em sua atividade gênica e examinando quais genes distinguiam cada cluster. Quando genes marcadores e Tangram concordavam, os rótulos eram aceitos; quando discordavam, os padrões gênicos locais no tecido tinham precedência. Finalmente, harmonizaram os resultados entre todas as lâminas em dez categorias amplas, como epitélio, vasos sanguíneos, grupos de células imunes, fibroblastos, melanócitos e um grupo genérico “outros”. Um patologista inspecionou visualmente os resultados nas imagens H&E para garantir que os rótulos fizessem sentido biológico.
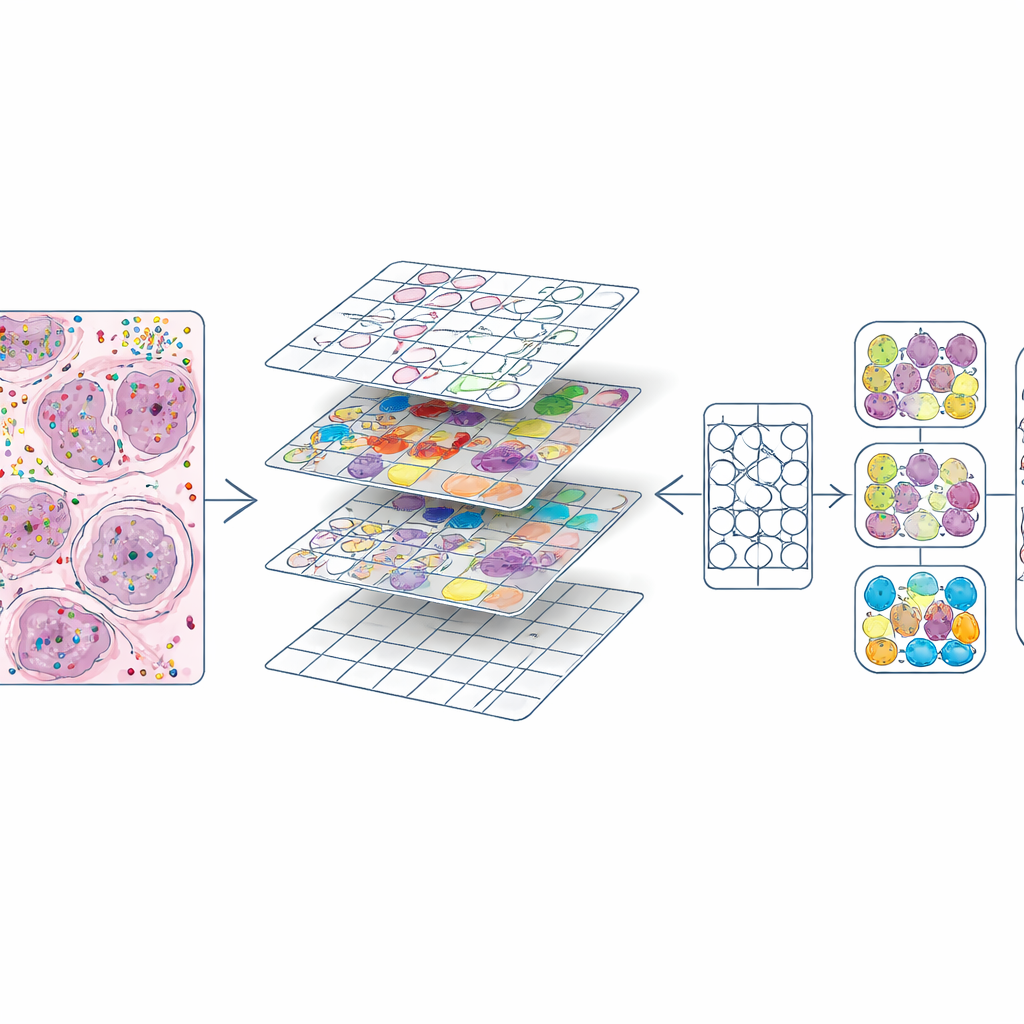
Transformando mapas moleculares ricos em material de treinamento
Uma vez que cada célula possuía posição e tipo, os autores cortaram cada lâmina H&E em pequenos quadrados, semelhantes a tiles de imagem em um programa de edição. Para cada tile produziram dois ingredientes principais: uma máscara delineando cada núcleo e um código de cor indicando seu tipo celular. Isso gerou cerca de 587.000 tiles em alta ampliação e um conjunto ligeiramente menor em baixa ampliação. Eles também compararam seus contornos de núcleos com os produzidos automaticamente por um modelo de aprendizado profundo existente (CellViT) e calcularam escores de concordância, permitindo que futuros usuários filtrem regiões de qualidade inferior. Todas essas informações — imagens, máscaras, contagens gênicas, rótulos celulares e escores de qualidade — são empacotadas em objetos de dados padronizados para que pesquisadores possam explorar ou reutilizar facilmente o conjunto.
Mostrando que computadores podem aprender com o novo atlas
Para demonstrar o que o STHELAR possibilita, a equipe ajustou finamente o modelo CellViT, um poderoso transformer de visão projetado para segmentar e classificar células em imagens H&E. Usando os rótulos do STHELAR como verdade de referência, treinaram o modelo para reconhecer nove classes celulares detalhadas e, em um segundo experimento, cinco grupos mais amplos (por exemplo, combinando vários tipos de células imunes). O modelo ajustado manteve bom desempenho na detecção e delineamento de núcleos e alcançou precisão satisfatória para células visualmente distintivas, como células epiteliais e melanócitos, enquanto subtipos imunes mais sutis permaneceram desafiadores. Eles também verificaram suas anotações contra um método de rotulagem independente (SingleR) e modelos alternativos baseados em RNA, encontrando, em geral, boa concordância.
O que isso significa para o diagnóstico do câncer no futuro
O STHELAR é menos um algoritmo único do que um atlas de referência: um elo aberto e em grande escala entre o que os patologistas veem em lâminas H&E padrão e o que a transcriptômica espacial revela sobre a atividade gênica em cada célula. Para não especialistas, a conclusão é que esse recurso facilita muito treinar e testar modelos computacionais capazes de inferir a composição celular de tumores diretamente a partir de imagens rotineiras, sem precisar realizar exames moleculares caros para cada paciente. À medida que esses modelos melhorarem, poderão ajudar médicos a ler a “conversa” molecular invisível dentro dos tumores a partir de lâminas comuns, apoiando diagnósticos mais precisos e tratamentos melhor ajustados.
Citação: Giraud-Sauveur, F., Blampey, Q., Benkirane, H. et al. STHELAR, a multi-tissue dataset linking spatial transcriptomics and histology for cell type annotation. Sci Data 13, 665 (2026). https://doi.org/10.1038/s41597-026-06937-6
Palavras-chave: microambiente tumoral, transcriptômica espacial, imagens histopatológicas, anotação de tipos celulares, aprendizado profundo em câncer