Clear Sky Science · it
STHELAR, un dataset multi-tessuto che collega trascrittomica spaziale e istologia per l’annotazione dei tipi cellulari
Perché guardare il cancro al microscopio non è sufficiente
I medici oncologi si affidano ancora in larga misura a ciò che vedono al microscopio: tessuto affettato sottile, colorato di rosa e viola con ematossilina ed eosina (H&E). Queste immagini rivelano forme e schemi, ma non possono mostrare direttamente quali geni sono attivi in ciascuna cellula. Una nuova risorsa chiamata STHELAR mette insieme questi due mondi, collegando la vista familiare dei vetrini istologici con misure “spaziali” all’avanguardia dell’attività genica. Per i lettori questo lavoro è importante perché apre la strada a strumenti più veloci ed economici che un giorno potrebbero leggere la composizione molecolare dei tumori a partire soltanto da immagini digitali ordinarie.
Vedere il tumore come un quartiere affollato
I tumori non sono solo cellule cancerose fuorvianti; sono quartieri affollati pieni di cellule immunitarie, vasi sanguigni, cellule di supporto e tessuto normale. La composizione e la disposizione di questi abitanti—il microambiente tumorale—possono determinare come un tumore cresce e come risponde alle terapie. La trascrittomica spaziale, una tecnologia recente, può mappare quali geni sono attivi in singole cellule preservandone la posizione precisa nel tessuto. Tuttavia, questi esperimenti sono costosi e tecnicamente impegnativi, quindi non fanno ancora parte dell’assistenza clinica di routine. Al contrario, le scansioni ad alta risoluzione dei vetrini H&E sono ormai comuni, economiche da conservare e già utilizzate in tutto il mondo. L’idea centrale di STHELAR è usare un numero limitato di esperimenti di trascrittomica spaziale come “insegnante” per milioni di cellule osservate nelle immagini H&E standard.
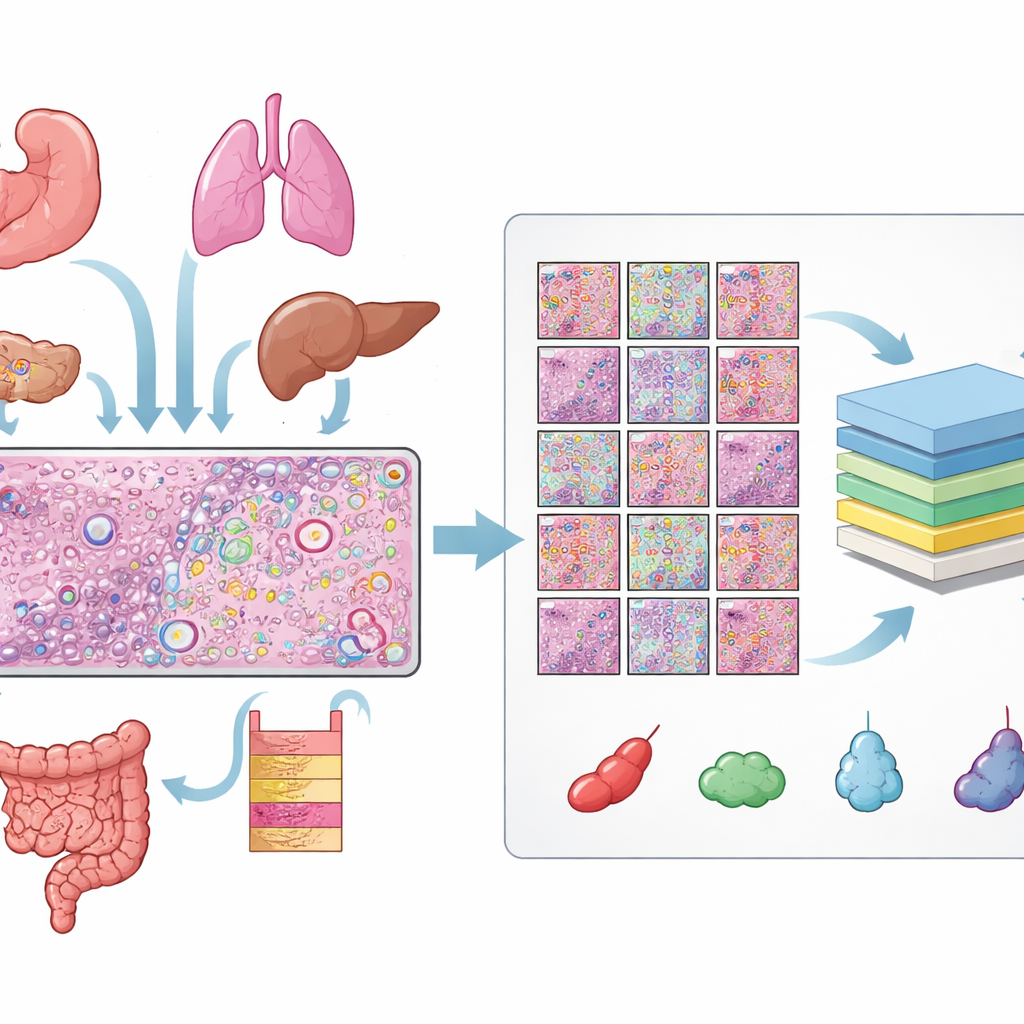
Costruire una gigantesca libreria di cellule etichettate
Gli autori hanno raccolto dati da 31 sezioni tissutali misurate con la piattaforma di trascrittomica spaziale Xenium di 10x Genomics, coprendo 16 tipi di tessuto umano e 22 campioni tumorali e 9 non tumorali. Per ogni sezione disponevano di tre viste corrispondenti: un vetrino H&E, un’immagine fluorescente che mostra i nuclei cellulari e una mappa delle singole molecole di RNA. Allineare queste viste ha richiesto controlli di qualità accurati e, per molti vetrini, un aggiustamento manuale affinché ogni nucleo nell’immagine fluorescente coincidesse con la corrispondente struttura nel vetrino H&E. Dalle immagini allineate hanno ottenuto oltre 11 milioni di cellule distinte e più di mezzo milione di piccoli riquadri H&E, ciascuno con contorni precisi di ogni nucleo.
Insegnare al computer cos’è ogni tipo cellulare
Sapere dove si trova ogni cellula non basta; il passo cruciale è decidere che tipo di cellula sia. Per farlo, il gruppo ha combinato la trascrittomica spaziale con grandi cataloghi esistenti di profili di RNA a cellula singola. Usando un metodo chiamato Tangram, hanno prima trasferito le probabili identità cellulari da questi atlanti di riferimento sui dati spaziali. Hanno poi migliorato queste etichette preliminari raggruppando le cellule in cluster in base all’attività genica ed esaminando quali geni distinguevano ciascun cluster. Quando i geni marker e Tangram concordavano, le etichette venivano accettate; quando non concordavano, la distribuzione locale dei geni nel tessuto aveva la precedenza. Infine, hanno armonizzato i risultati su tutti i vetrini in dieci categorie ampie come epiteliali, vasi sanguigni, gruppi di cellule immunitarie, fibroblasti, melanociti e un gruppo generico “altro”. Un patologo ha ispezionato visivamente i risultati sulle immagini H&E per assicurarsi che le etichette avessero senso biologico.
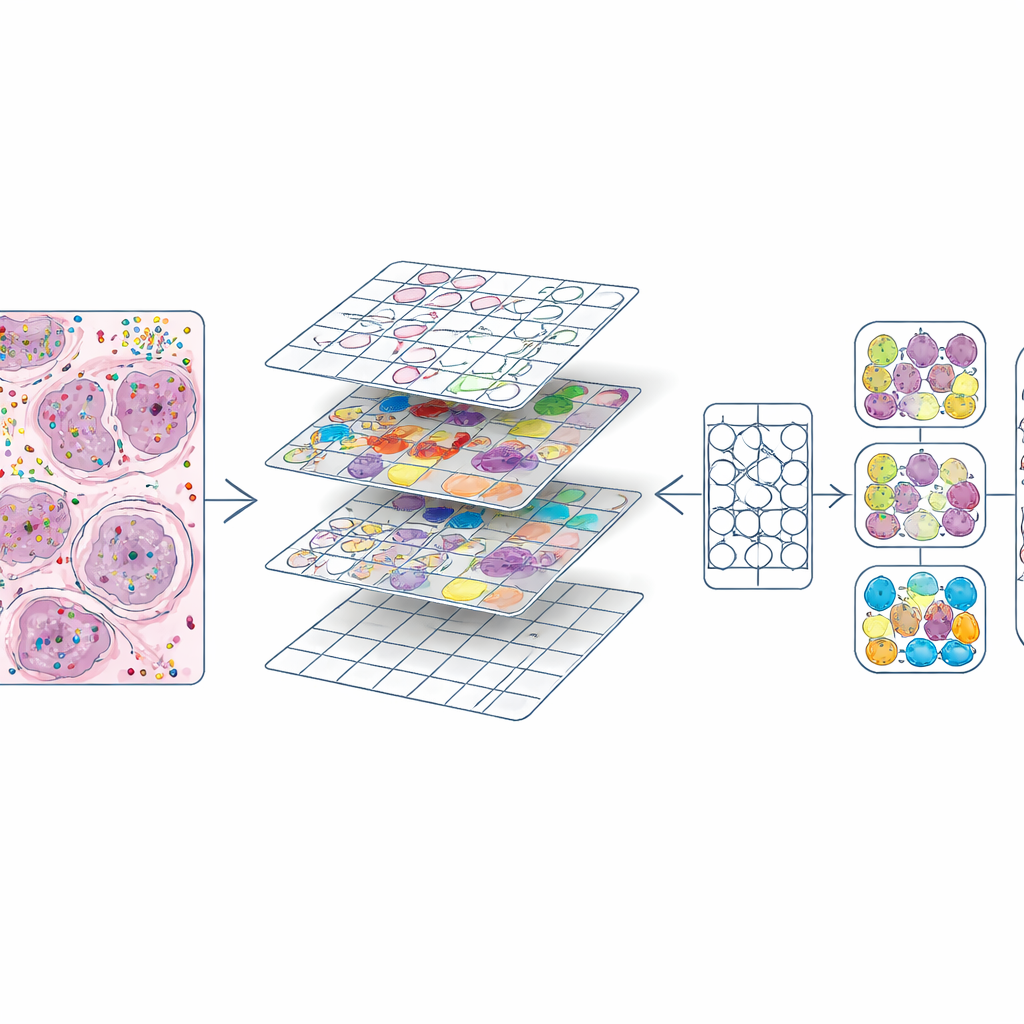
Trasformare mappe molecolari ricche in materiale di addestramento
Una volta che ogni cellula aveva sia una posizione sia un tipo, gli autori hanno ritagliato ogni vetrino H&E in piccoli quadrati, simili alle tessere di un programma di editing fotografico. Per ogni tessera hanno prodotto due ingredienti chiave: una maschera che delimita ogni nucleo e un codice colore che indica il suo tipo cellulare. Questo ha creato circa 587.000 tessere ad alta ingrandimento e un insieme leggermente più piccolo a ingrandimento inferiore. Hanno anche confrontato i contorni dei nuclei con quelli prodotti automaticamente da un modello di deep learning esistente (CellViT) e calcolato punteggi di concordanza, permettendo agli utenti futuri di filtrare le regioni di qualità inferiore. Tutte queste informazioni—immagini, maschere, conteggi genici, etichette cellulari e punteggi di qualità—sono confezionate in oggetti di dati standardizzati in modo che i ricercatori possano facilmente esplorare o riutilizzare il dataset.
Dimostrare che i computer possono imparare dal nuovo atlante
Per dimostrare cosa permette STHELAR, il team ha effettuato un fine-tuning del modello CellViT, un potente vision transformer progettato per segmentare e classificare le cellule nelle immagini H&E. Usando le etichette di STHELAR come verità di riferimento, hanno addestrato il modello a riconoscere nove classi cellulari dettagliate e, in un secondo esperimento, cinque gruppi più ampi (ad esempio combinando diversi tipi di cellule immunitarie). Il modello messo a punto ha mantenuto buone prestazioni nel rilevare e delimitare i nuclei e ha ottenuto buona accuratezza per cellule visivamente distintive come le cellule epiteliali e i melanociti, mentre sottotipi immunitari più sottili sono rimasti una sfida. Hanno inoltre confrontato le loro annotazioni con un metodo di etichettatura indipendente (SingleR) e con modelli alternativi basati sull’RNA, riscontrando in generale buona concordanza.
Cosa significa per la diagnosi del cancro in futuro
STHELAR è meno un singolo algoritmo che un atlante di riferimento: un collegamento aperto e su larga scala tra ciò che i patologi vedono sui vetrini H&E standard e ciò che la trascrittomica spaziale rivela sull’attività genica in ogni cellula. Per i non specialisti, la conclusione è che questa risorsa rende molto più semplice addestrare e testare modelli computazionali in grado di inferire la composizione cellulare dei tumori direttamente dalle immagini di routine, senza dover eseguire costosi saggi molecolari per ogni paziente. Man mano che questi modelli migliorano, potrebbero aiutare i medici a leggere la “conversazione” molecolare invisibile all’interno dei tumori a partire da vetrini ordinari, supportando diagnosi più precise e terapie meglio mirate.
Citazione: Giraud-Sauveur, F., Blampey, Q., Benkirane, H. et al. STHELAR, a multi-tissue dataset linking spatial transcriptomics and histology for cell type annotation. Sci Data 13, 665 (2026). https://doi.org/10.1038/s41597-026-06937-6
Parole chiave: microambiente tumorale, trascrittomica spaziale, imaging istopatologico, annotazione dei tipi cellulari, deep learning nel cancro