Clear Sky Science · fr
STHELAR, un jeu de données multi-tissus liant la transcriptomique spatiale et l'histologie pour l'annotation des types cellulaires
Pourquoi regarder le cancer au microscope ne suffit pas
Les oncologues se fient encore largement à ce qu'ils observent au microscope : des tranches de tissus mince teintées en rose et violet avec l'hématoxyline et l'éosine (H&E). Ces images révèlent des formes et des motifs, mais elles ne montrent pas directement quels gènes sont actifs dans chaque cellule. Une nouvelle ressource nommée STHELAR relie ces deux approches, associant la vue familière des lames de tissu aux mesures « spatiales » de l'activité génique. Pour le lecteur, cette avancée est importante parce qu'elle ouvre la voie à des outils plus rapides et moins coûteux qui, un jour, pourraient lire la composition moléculaire des tumeurs à partir d'images numériques ordinaires.
Voir la tumeur comme un quartier dense
Les tumeurs ne se résument pas à des cellules cancéreuses isolées ; ce sont des quartiers densément peuplés remplis de cellules immunitaires, de vaisseaux sanguins, de cellules de soutien et de tissu normal. La composition et l'organisation de ces habitants — le microenvironnement tumoral — peuvent influencer la croissance du cancer et sa réponse au traitement. La transcriptomique spatiale, une technologie récente, permet de cartographier les gènes exprimés dans des cellules individuelles tout en préservant leur position précise dans le tissu. Cependant, ces expériences sont coûteuses et techniquement exigeantes, et ne font pas encore partie des soins de routine. En revanche, les numérisations haute résolution des lames H&E sont désormais courantes, peu coûteuses à stocker et utilisées dans le monde entier. L'idée centrale de STHELAR est d'utiliser un nombre limité d'expériences de transcriptomique spatiale comme « professeur » pour des millions de cellules visibles sur des images H&E standard.
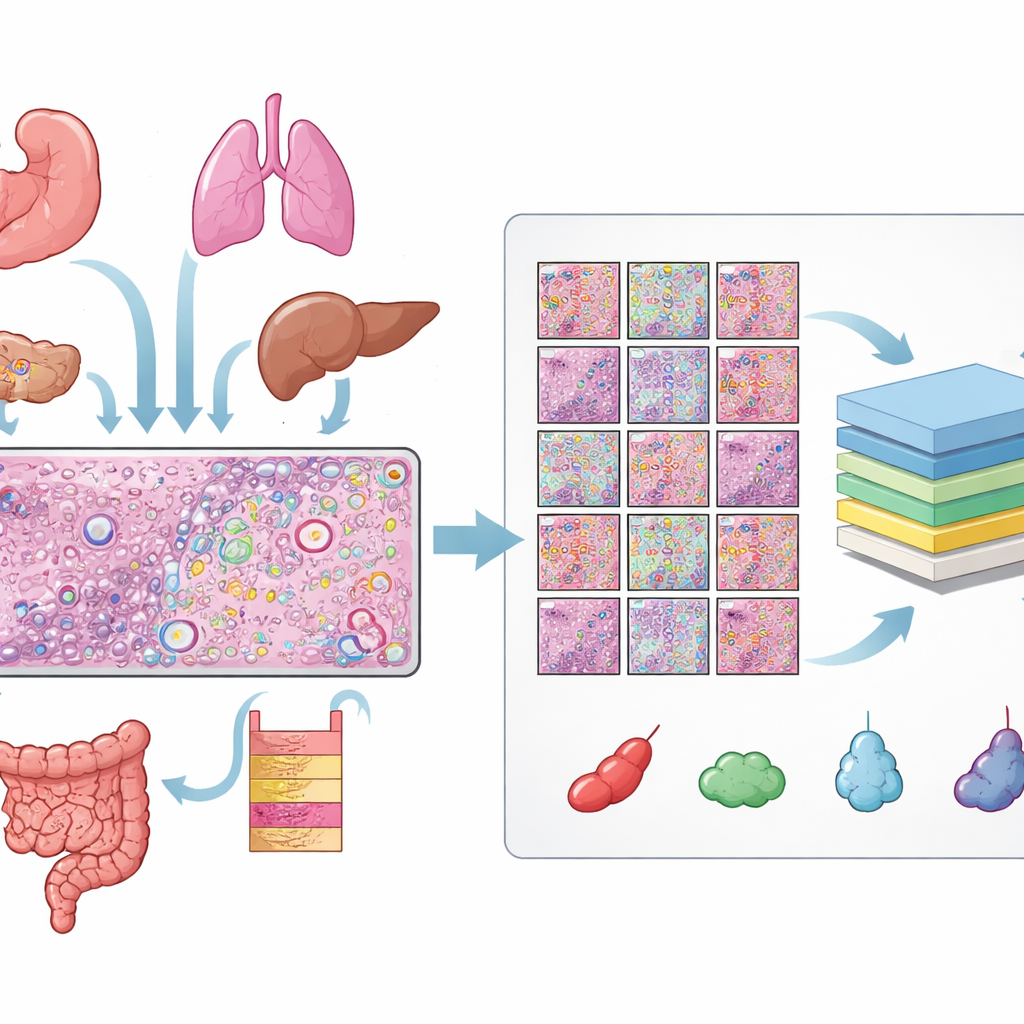
Construire une vaste bibliothèque de cellules annotées
Les auteurs ont rassemblé des données provenant de 31 coupes de tissu mesurées avec la plateforme de transcriptomique spatiale Xenium de 10x Genomics, couvrant 16 types de tissus humains et 22 échantillons cancéreux et 9 non cancéreux. Pour chaque coupe, ils disposaient de trois vues appariées : une lame H&E, une image fluorescente montrant les noyaux cellulaires et une carte des molécules d'ARN individuelles. L'alignement de ces vues a exigé des contrôles qualité rigoureux et, pour de nombreuses lames, un ajustement manuel afin que chaque noyau visible en fluorescence corresponde à la structure correspondante sur la lame H&E. À partir de ces images alignées, ils ont obtenu plus de 11 millions de cellules distinctes et plus d’un demi-million de petits patchs H&E, chacun avec les contours précis de chaque noyau.
Apprendre à l'ordinateur à reconnaître chaque type cellulaire
Savoir où se trouve chaque cellule ne suffit pas ; l'étape cruciale est de déterminer de quel type de cellule il s'agit. Pour cela, l'équipe a combiné la transcriptomique spatiale avec de grands catalogues existants de profils d'ARN unicellulaire. En utilisant une méthode appelée Tangram, ils ont d'abord transféré des identités cellulaires probables depuis ces atlas de référence vers les données spatiales. Ils ont ensuite affiné ces étiquettes préliminaires en regroupant les cellules en clusters selon leur activité génique et en examinant les gènes qui distinguaient chaque groupe. Lorsque les gènes marqueurs et Tangram concordaient, les étiquettes étaient acceptées ; en cas de désaccord, les motifs géniques locaux dans le tissu prévalaient. Enfin, ils ont harmonisé les résultats sur toutes les lames en dix catégories larges telles qu'épithélial, vaisseau sanguin, groupes de cellules immunitaires, fibroblastes, mélanocytes et un groupe fourre‑tout « autre ». Un pathologiste a inspecté visuellement les résultats sur les images H&E pour s'assurer que les étiquettes avaient un sens biologique.
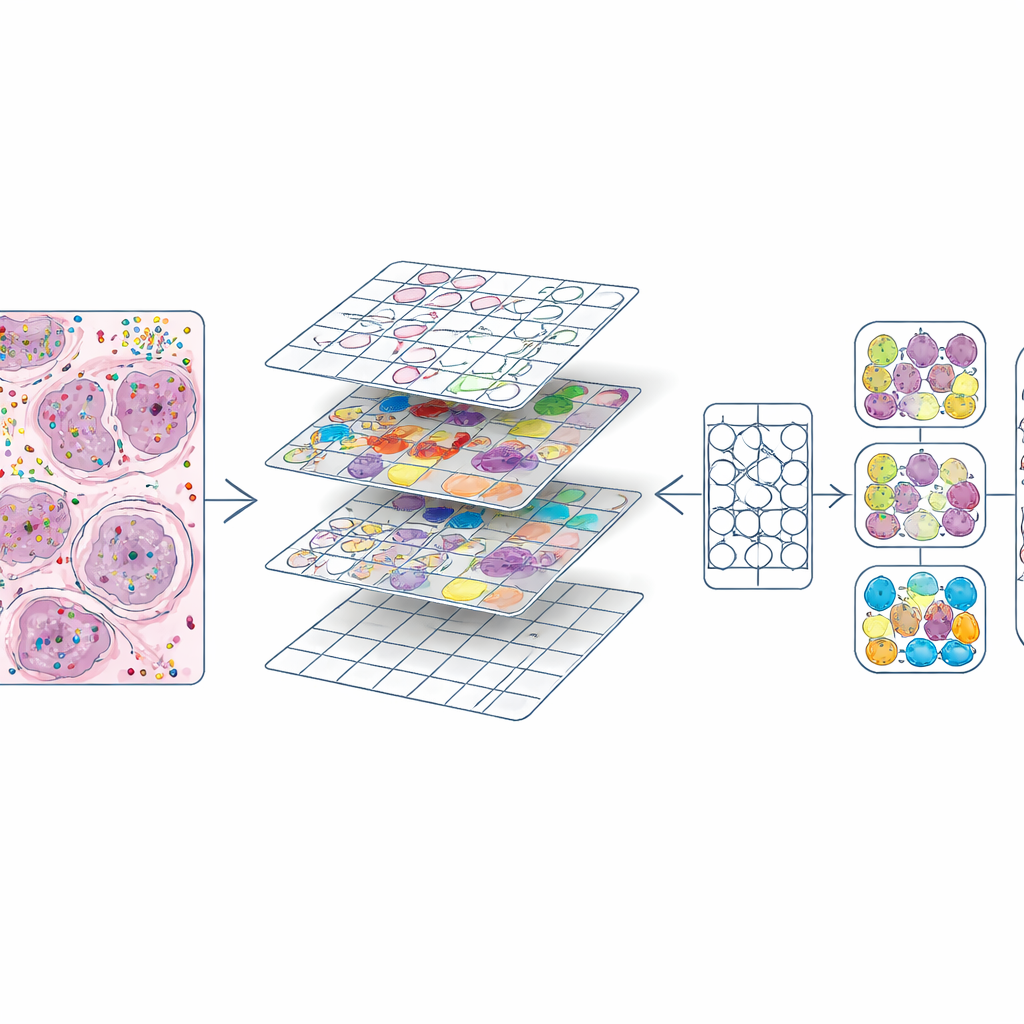
Transformer les cartes moléculaires riches en matériel d'entraînement
Une fois que chaque cellule avait à la fois une position et un type, les auteurs ont découpé chaque lame H&E en petits carrés, similaires aux tuiles d'image dans un logiciel de retouche photo. Pour chaque tuile, ils ont produit deux éléments clés : un masque délimitant chaque noyau et un code couleur indiquant son type cellulaire. Cela a créé environ 587 000 tuiles à fort grossissement et un ensemble légèrement plus petit à un grossissement inférieur. Ils ont aussi comparé leurs contours de noyaux à ceux générés automatiquement par un modèle d'apprentissage profond existant (CellViT) et calculé des scores d'accord, permettant aux futurs utilisateurs d'exclure les régions de moindre qualité. Toutes ces informations — images, masques, comptes de gènes, étiquettes cellulaires et scores de qualité — sont regroupées dans des objets de données standardisés afin que les chercheurs puissent facilement explorer ou réutiliser le jeu de données.
Montrer que les ordinateurs peuvent apprendre à partir du nouvel atlas
Pour démontrer ce que permet STHELAR, l'équipe a affiné le modèle CellViT, un puissant transformeur de vision conçu pour segmenter et classer les cellules dans les images H&E. En utilisant les étiquettes de STHELAR comme vérité terrain, ils ont entraîné le modèle à reconnaître neuf classes cellulaires détaillées et, dans une seconde expérience, cinq groupes plus larges (par exemple en combinant plusieurs types de cellules immunitaires). Le modèle affiné a conservé de bonnes performances pour détecter et délimiter les noyaux et a obtenu une bonne précision pour les cellules visuellement distinctes comme les cellules épithéliales et les mélanocytes, tandis que des sous‑types immunitaires plus subtils restaient difficiles. Ils ont également comparé leurs annotations à une méthode d'étiquetage indépendante (SingleR) et à des modèles alternatifs basés sur l'ARN, trouvant généralement une bonne concordance.
Ce que cela signifie pour le diagnostic du cancer à l'avenir
STHELAR est moins un algorithme unique qu'un atlas de référence : un lien ouvert et à grande échelle entre ce que les pathologistes voient sur des lames H&E standard et ce que la transcriptomique spatiale révèle sur l'activité génique dans chaque cellule. Pour les non‑spécialistes, l'essentiel est que cette ressource facilite grandement l'entraînement et l'évaluation de modèles informatiques capables d'inférer la composition cellulaire des tumeurs directement à partir d'images de routine, sans devoir réaliser des tests moléculaires coûteux pour chaque patient. À mesure que ces modèles s'amélioreront, ils pourraient aider les médecins à lire la « conversation » moléculaire invisible au sein des tumeurs à partir de lames ordinaires, soutenant des diagnostics plus précis et des traitements mieux adaptés.
Citation: Giraud-Sauveur, F., Blampey, Q., Benkirane, H. et al. STHELAR, a multi-tissue dataset linking spatial transcriptomics and histology for cell type annotation. Sci Data 13, 665 (2026). https://doi.org/10.1038/s41597-026-06937-6
Mots-clés: microenvironnement tumoral, transcriptomique spatiale, imagerie histopathologique, annotation des types cellulaires, apprentissage profond en cancérologie