Clear Sky Science · es
STHELAR, un conjunto de datos multitisular que vincula transcriptómica espacial e histología para la anotación de tipos celulares
Por qué mirar el cáncer al microscopio no es suficiente
Los oncólogos siguen confiando en gran medida en lo que ven al microscopio: tejido finamente cortado, teñido de rosa y púrpura con hematoxilina y eosina (H&E). Estas imágenes revelan formas y patrones, pero no pueden mostrar directamente qué genes están activos en cada célula. Un nuevo recurso llamado STHELAR une estos dos mundos, vinculando la vista familiar de los cortes histológicos con medidas espaciales de vanguardia sobre la actividad génica. Para los lectores, este trabajo importa porque abre la puerta a herramientas más rápidas y económicas que algún día podrían leer la composición molecular de los tumores a partir de imágenes digitales ordinarias.
Ver el tumor como un vecindario abarrotado
Los tumores son más que células cancerosas descontroladas; son vecindarios concurridos llenos de células inmunitarias, vasos sanguíneos, células de soporte y tejido normal. La mezcla y disposición de estos habitantes —el microambiente tumoral— puede influir en cómo crece un cáncer y cómo responde al tratamiento. La transcriptómica espacial, una tecnología reciente, puede mapear qué genes están activados en células individuales manteniendo su posición exacta en el tejido. Sin embargo, estos experimentos son costosos y técnicamente exigentes, por lo que aún no forman parte de la atención rutinaria. En contraste, los escaneos de alta resolución de cortes H&E son ahora comunes, baratos de almacenar y ya se usan en todo el mundo. La idea central detrás de STHELAR es usar un conjunto limitado de experimentos de transcriptómica espacial como "maestro" para millones de células visibles en imágenes H&E estándar.
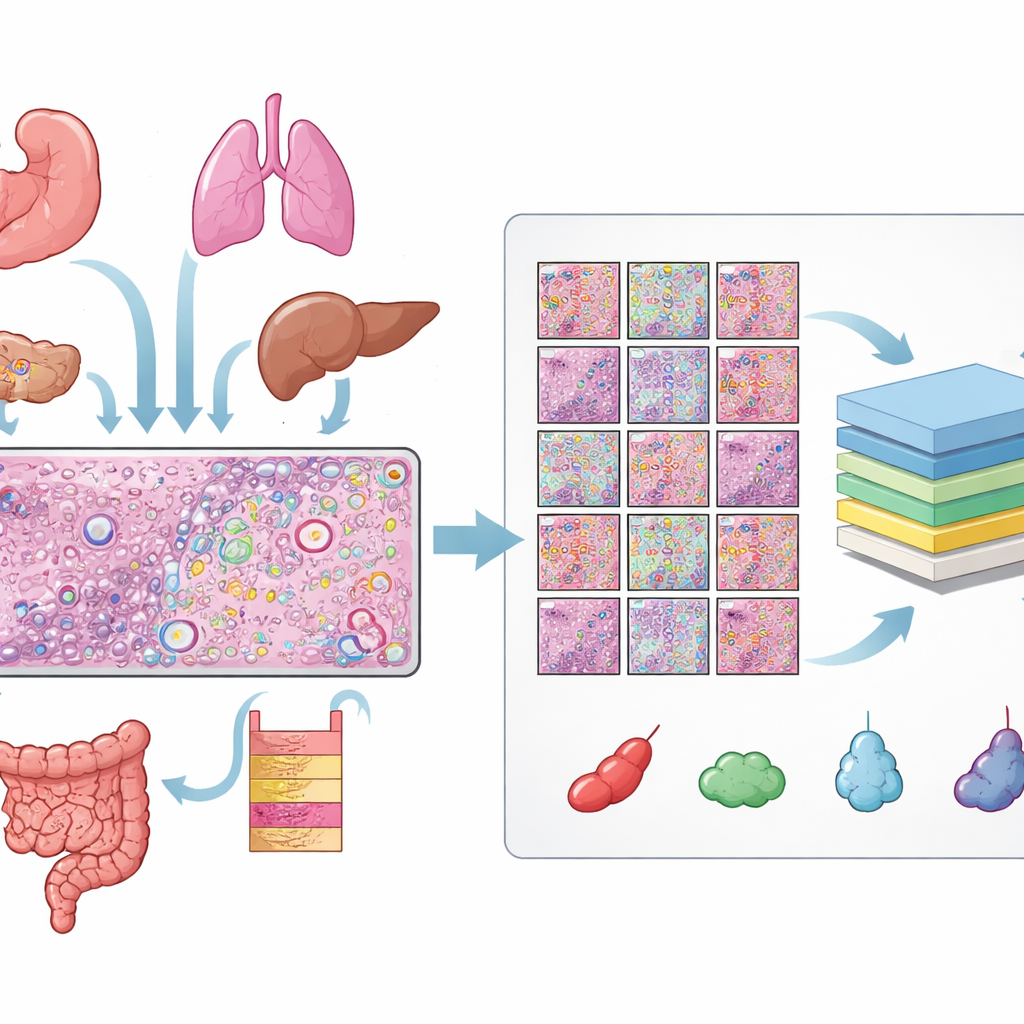
Construir una enorme biblioteca de células etiquetadas
Los autores reunieron datos de 31 secciones de tejido medidas con la plataforma de transcriptómica espacial Xenium de 10x Genomics, abarcando 16 tipos de tejido humano y 22 muestras tumorales y 9 no tumorales. Para cada sección dispusieron de tres vistas emparejadas: un corte H&E, una imagen fluorescente que muestra los núcleos celulares y un mapa de moléculas individuales de ARN. Alinear estas vistas requirió controles de calidad cuidadosos y, para muchas láminas, un ajuste fino manual para que cada núcleo en la imagen fluorescente coincidiera con la estructura correspondiente en el corte H&E. A partir de estas imágenes alineadas obtuvieron más de 11 millones de células distintas y más de medio millón de pequeños fragmentos (patches) H&E, cada uno con contornos precisos de cada núcleo.
Enseñar a la computadora qué tipo de célula es cada una
Saber dónde se encuentra cada célula no es suficiente; el paso crucial es decidir qué tipo de célula es. Para ello, el equipo combinó la transcriptómica espacial con grandes catálogos existentes de perfiles de ARN de célula única. Usando un método llamado Tangram, primero transfirieron las identidades celulares probables de estos atlas de referencia a los datos espaciales. Luego mejoraron estas etiquetas preliminares agrupando células en clústeres según su actividad génica y examinando qué genes distinguían cada grupo. Cuando los genes marcadores y Tangram coincidían, se aceptaban las etiquetas; cuando discrepaban, prevalecían los patrones locales de expresión génica en el tejido. Finalmente, armonizaron los resultados entre todas las láminas en diez categorías generales como epitelial, vaso sanguíneo, grupos de células inmunitarias, fibroblastos, melanocitos y un grupo residual "otros". Un patólogo inspeccionó visualmente los resultados sobre las imágenes H&E para asegurar que las etiquetas tuvieran sentido biológico.
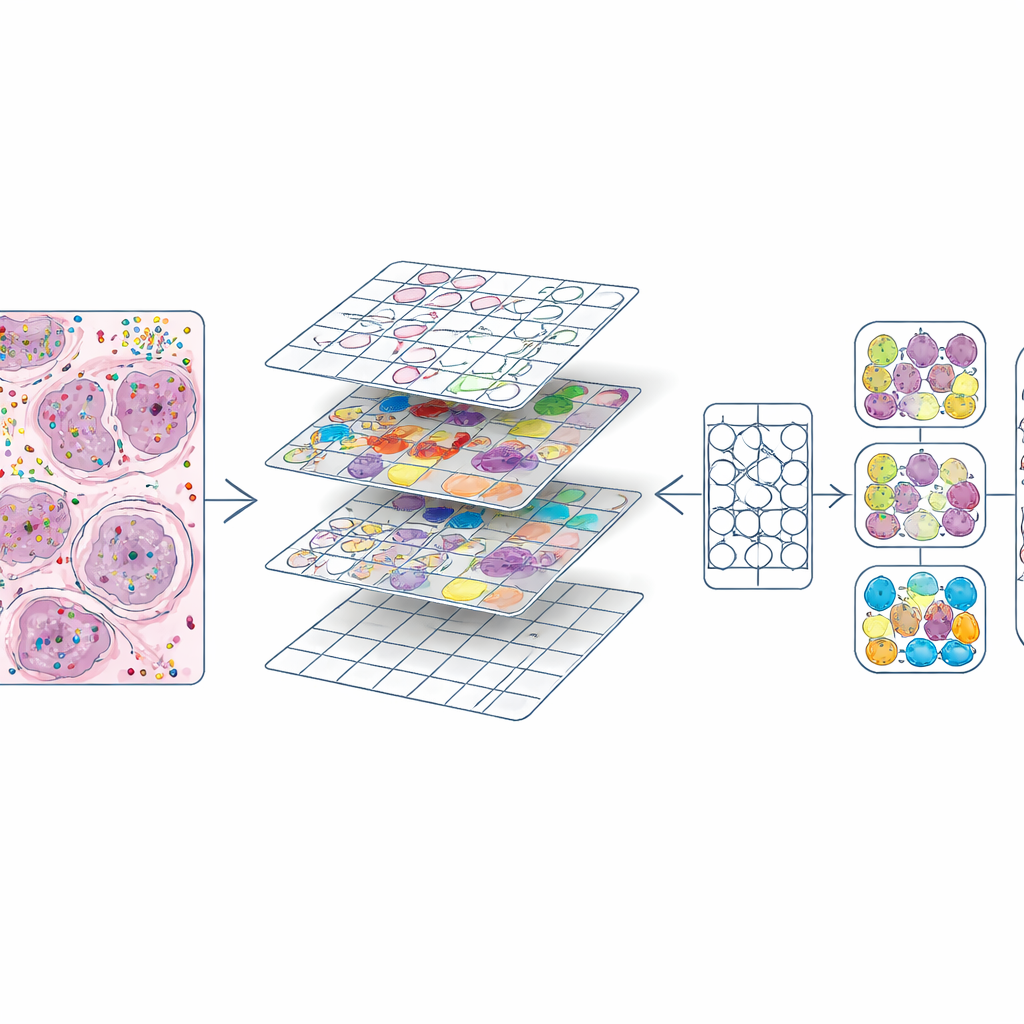
Convertir mapas moleculares ricos en material de entrenamiento
Una vez que cada célula tuvo tanto posición como tipo, los autores cortaron cada lámina H&E en pequeños cuadrados, similar a los mosaicos de imágenes en un programa de edición fotográfica. Para cada mosaico produjeron dos ingredientes clave: una máscara que delineaba cada núcleo y un código de color que indicaba su tipo celular. Esto creó cerca de 587.000 mosaicos a alta ampliación y un conjunto algo menor a menor ampliación. También compararon sus contornos de núcleos con los producidos automáticamente por un modelo de aprendizaje profundo existente (CellViT) y calcularon puntuaciones de concordancia, lo que permitirá a futuros usuarios filtrar regiones de menor calidad. Toda esta información —imágenes, máscaras, recuentos génicos, etiquetas celulares y puntuaciones de calidad— se agrupa en objetos de datos estandarizados para que los investigadores puedan explorar o reutilizar fácilmente el conjunto de datos.
Demostrar que las computadoras pueden aprender del nuevo atlas
Para demostrar lo que permite STHELAR, el equipo ajustó finamente el modelo CellViT, un potente transformer de visión diseñado para segmentar y clasificar células en imágenes H&E. Usando las etiquetas de STHELAR como verdad de referencia, entrenaron el modelo para reconocer nueve clases celulares detalladas y, en un segundo experimento, cinco grupos más amplios (por ejemplo, combinando varios tipos de células inmunitarias). El modelo ajustado mantuvo un buen rendimiento al detectar y delinear núcleos y alcanzó buena precisión para células visualmente distintivas como las epiteliales y los melanocitos, mientras que subtipos inmunitarios más sutiles siguieron siendo un desafío. También verificaron sus anotaciones frente a un método de etiquetado independiente (SingleR) y modelos alternativos basados en ARN, encontrando en general buena concordancia.
Qué significa esto para el diagnóstico futuro del cáncer
STHELAR es menos un único algoritmo que un atlas de referencia: un enlace abierto y a gran escala entre lo que los patólogos ven en cortes H&E estándar y lo que la transcriptómica espacial revela sobre la actividad génica en cada célula. Para no especialistas, la conclusión es que este recurso facilita enormemente entrenar y evaluar modelos informáticos que pueden inferir la composición celular de los tumores directamente desde imágenes rutinarias, sin tener que realizar ensayos moleculares costosos para cada paciente. A medida que estos modelos mejoren, podrían ayudar a los médicos a leer la "conversación" molecular invisible dentro de los tumores a partir de cortes ordinarios, respaldando diagnósticos más precisos y tratamientos mejor adaptados.
Cita: Giraud-Sauveur, F., Blampey, Q., Benkirane, H. et al. STHELAR, a multi-tissue dataset linking spatial transcriptomics and histology for cell type annotation. Sci Data 13, 665 (2026). https://doi.org/10.1038/s41597-026-06937-6
Palabras clave: microambiente tumoral, transcriptómica espacial, imágenes histopatológicas, anotación de tipos celulares, aprendizaje profundo en cáncer