Clear Sky Science · de
STHELAR, ein Multi-Gewebe-Datensatz, der räumliche Transkriptomik und Histologie zur Zelltyp-Annotation verbindet
Warum der Blick durch das Mikroskop allein nicht ausreicht
Krebsärzte verlassen sich nach wie vor stark auf das, was sie im Mikroskop sehen: dünn aufgeschnittenes, pink‑ und violettgefärbtes Gewebe, das mit Hämatoxylin und Eosin (H&E) gefärbt wurde. Diese Bilder zeigen Formen und Muster, können aber nicht direkt offenlegen, welche Gene in jeder einzelnen Zelle aktiv sind. Eine neue Ressource namens STHELAR verbindet diese beiden Welten und verknüpft die vertraute Ansicht von Gewebeschnitten mit modernen „räumlichen“ Messungen der Genaktivität. Für Leser ist diese Arbeit wichtig, weil sie den Weg zu schnelleren, günstigeren Werkzeugen ebnet, die eines Tages allein aus gewöhnlichen digitalen Bildern die molekulare Zusammensetzung von Tumoren ablesen könnten.
Den Tumor als dichtes Viertel sehen
Tumoren bestehen nicht nur aus entarteten Krebszellen; sie sind dicht besiedelte Viertel voller Immunzellen, Blutgefäße, Stütz- und Normalgewebe. Das Sortiment und die Anordnung dieser Bewohner – die Tumormikroumgebung – können beeinflussen, wie ein Krebs wächst und wie er auf Therapien reagiert. Räumliche Transkriptomik, eine neuere Technologie, kann abbilden, welche Gene in einzelnen Zellen an- oder abgeschaltet sind und dabei deren exakte Position im Gewebe erhalten. Diese Experimente sind jedoch kosten- und technisch aufwändig und noch nicht Teil der routinemäßigen Versorgung. Im Gegensatz dazu sind hochaufgelöste Scans von H&E‑Präparaten mittlerweile verbreitet, billig zu speichern und weltweit im Einsatz. Die zentrale Idee von STHELAR ist, eine begrenzte Anzahl räumlicher Transkriptomik-Experimente als „Lehrer“ für Millionen von Zellen in standardmäßigen H&E‑Bildern zu verwenden.
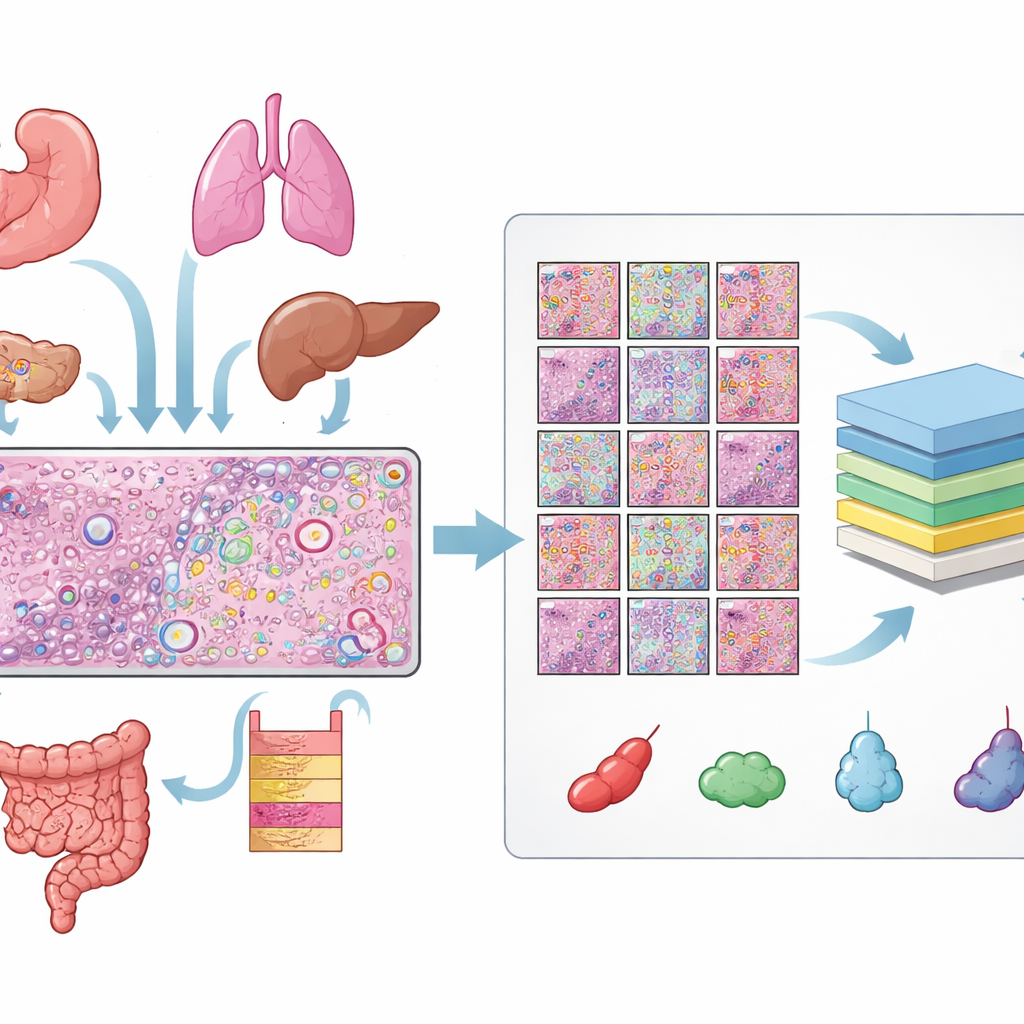
Aufbau einer riesigen Bibliothek beschrifteter Zellen
Die Autorinnen und Autoren sammelten Daten aus 31 Gewebeschnitten, die mit der Xenium‑Plattform von 10x Genomics räumlich-transkriptomisch gemessen wurden. Die Daten deckten 16 menschliche Gewebetypen sowie 22 Krebs- und 9 Nicht‑Krebs‑Proben ab. Zu jedem Schnitt lagen drei passende Ansichten vor: ein H&E‑Präparat, ein Fluoreszenzbild, das Zellkerne zeigt, und eine Karte einzelner RNA‑Moleküle. Das Ausrichten dieser Ansichten erforderte sorgfältige Qualitätskontrollen und bei vielen Präparaten manuelle Feinanpassungen, damit jeder Kern im Fluoreszenzbild mit der entsprechenden Struktur im H&E‑Bild übereinstimmte. Aus diesen ausgerichteten Bildern gewannen sie über 11 Millionen einzelne Zellen und mehr als eine halbe Million kleiner H&E‑Ausschnitte, jeweils mit präzisen Umrissen jeder einzelnen Zellkernstruktur.
Dem Computer beibringen, welche Zelltypen es gibt
Zu wissen, wo sich jede Zelle befindet, reicht nicht aus; der entscheidende Schritt ist, festzustellen, um welchen Zelltyp es sich handelt. Dafür kombinierte das Team räumliche Transkriptomik mit großen vorhandenen Katalogen von Einzelzell-RNA‑Profilen. Mit einer Methode namens Tangram übertrugen sie zunächst wahrscheinliche Zellidentitäten aus diesen Referenzatlanten auf die räumlichen Daten. Anschließend verbesserten sie diese vorläufigen Beschriftungen, indem sie Zellen anhand ihrer Genaktivität in Cluster gruppierten und untersuchten, welche Gene die einzelnen Cluster unterschieden. Wenn Marker‑Gene und Tangram übereinstimmten, wurden die Labels übernommen; bei Widersprüchen hatten die lokalen Genmuster im Gewebe Vorrang. Abschließend harmonisierten sie die Ergebnisse über alle Schnitte hinweg zu zehn breiten Kategorien, etwa Epithelzellen, Blutgefäßzellen, Gruppen von Immunzellen, Fibroblasten, Melanozyten und einer Sammelgruppe „Sonstige“. Eine Pathologin bzw. ein Pathologe überprüfte die Resultate visuell in den H&E‑Bildern, um sicherzustellen, dass die Zuordnungen biologisch sinnvoll waren.
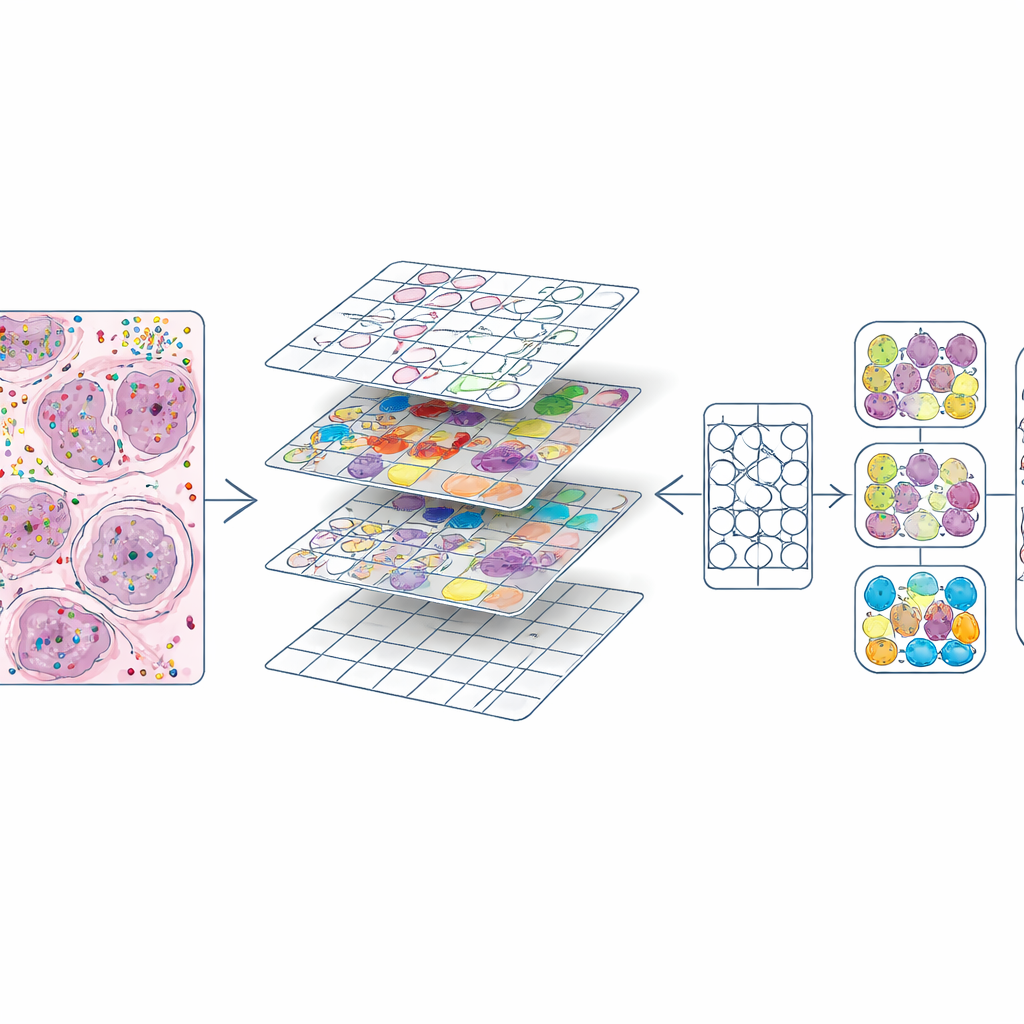
Reiche molekulare Karten in Trainingsmaterial verwandeln
Sobald jede Zelle sowohl eine Position als auch einen Typ hatte, schnitten die Autorinnen und Autoren jede H&E‑Probe in kleine Quadrate, ähnlich Bildkacheln in einem Bildbearbeitungsprogramm. Für jede Kachel erzeugten sie zwei zentrale Komponenten: eine Maske, die jeden Zellkern umreißt, und einen Farbcode, der den Zelltyp angibt. So entstanden etwa 587.000 Kacheln bei hoher Vergrößerung und eine etwas kleinere Menge bei niedrigerer Vergrößerung. Sie verglichen außerdem ihre Kernumrisse mit denen, die automatisch von einem bestehenden Deep‑Learning‑Modell (CellViT) erzeugt wurden, und berechneten Übereinstimmungswerte, sodass künftige Nutzer minderwertige Bereiche herausfiltern können. All diese Informationen – Bilder, Masken, Genzählungen, Zelllabels und Qualitätswerte – sind in standardisierten Datenobjekten gebündelt, damit Forschende den Datensatz leicht erkunden oder wiederverwenden können.
Zeigen, dass Computer aus dem neuen Atlas lernen können
Um zu demonstrieren, was STHELAR ermöglicht, verfeinerte das Team das CellViT‑Modell, einen leistungsfähigen Vision Transformer zum Segmentieren und Klassifizieren von Zellen in H&E‑Bildern. Mit den Labels von STHELAR als Ground Truth trainierten sie das Modell, neun detaillierte Zellklassen zu erkennen und in einem zweiten Experiment fünf breitere Gruppen (etwa durch Zusammenfassen mehrerer Immunzelltypen). Das feinabgestimmte Modell behielt eine starke Leistung beim Erkennen und Umreißen von Zellkernen und erreichte gute Genauigkeit für visuell markante Zellen wie Epithelzellen und Melanozyten, während subtilere Immununtertypen weiterhin herausfordernd blieben. Sie verglichen ihre Annotationen zudem mit einer unabhängigen Kennzeichnungs‑Methode (SingleR) und alternativen RNA‑basierten Modellen und fanden insgesamt gute Übereinstimmung.
Was das für die künftige Krebsdiagnostik bedeutet
STHELAR ist weniger ein einzelner Algorithmus als ein Referenzatlas: eine offene, groß angelegte Verbindung zwischen dem, was Pathologinnen und Pathologen auf standardmäßigen H&E‑Präparaten sehen, und dem, was räumliche Transkriptomik über die Genaktivität in jeder Zelle offenbart. Für Nicht‑Spezialisten lautet die Quintessenz, dass diese Ressource das Trainieren und Testen von Computermodellen erheblich erleichtert, die die zelluläre Zusammensetzung von Tumoren direkt aus Routinebildern ableiten können, ohne für jede Patientin bzw. jeden Patienten teure molekulare Tests durchführen zu müssen. Wenn sich solche Modelle verbessern, könnten sie helfen, die unsichtbare molekulare „Konversation“ innerhalb von Tumoren aus gewöhnlichen Präparaten zu lesen und so präzisere Diagnosen und besser abgestimmte Behandlungen zu unterstützen.
Zitation: Giraud-Sauveur, F., Blampey, Q., Benkirane, H. et al. STHELAR, a multi-tissue dataset linking spatial transcriptomics and histology for cell type annotation. Sci Data 13, 665 (2026). https://doi.org/10.1038/s41597-026-06937-6
Schlüsselwörter: Tumormikroumgebung, räumliche Transkriptomik, histopathologische Bildgebung, Zelltyp-Annotation, Deep Learning bei Krebs