Clear Sky Science · pt
Um canal TRP heteromérico que funciona como um receptor acoplado à proteína G ativado por WNT
Por que uma proteína renal importa para todos
A doença renal policística autossômica dominante (DRPAD) é um dos distúrbios renais hereditários mais comuns e uma das principais causas de insuficiência renal no mundo. Há décadas, os cientistas conhecem os genes centrais envolvidos — PKD1 e PKD2 —, mas não o que seus produtos proteicos realmente fazem dentro das células. Este artigo revela que essas proteínas formam um receptor incomum na superfície celular que se comunica diretamente com interruptores clássicos de sinalização dentro da célula. Ao explicar como esse sistema normalmente ajuda a manter sob controle uma molécula mensageira chave, o trabalho oferece uma imagem mais clara de como os cistos surgem na DRPAD e aponta novas maneiras de tratar a doença.
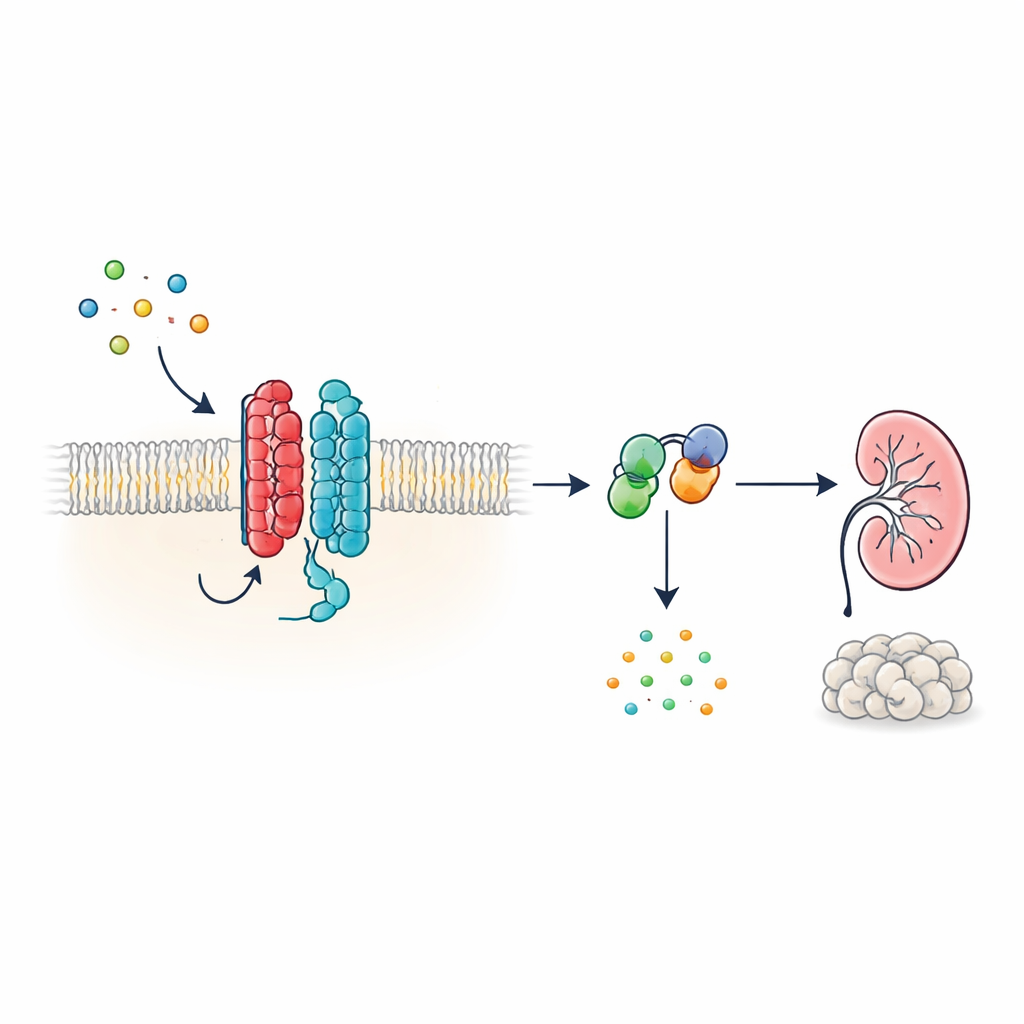
Um novo tipo de antena celular
Muitos hormônios e sinais no corpo atuam por meio de receptores acoplados à proteína G (GPCRs), uma enorme família de proteínas de superfície celular com uma estrutura característica de sete segmentos que atravessam a membrana. O PKD1, no entanto, parece diferente: tem 11 segmentos e foi agrupado com canais iônicos em vez de receptores clássicos. Os autores mostram que, apesar dessa forma não convencional, o PKD1 se comporta como um verdadeiro GPCR quando emparelhado com o PKD2. Juntas, as duas proteínas formam um complexo na membrana que responde a proteínas WNT — moléculas sinalizadoras versáteis envolvidas no desenvolvimento e na manutenção dos tecidos. Quando as WNT se ligam ao PKD1, o complexo ativa proteínas G intracelulares e altera os níveis de AMP cíclico (cAMP), uma pequena molécula que influencia fortemente o crescimento celular e a secreção de fluidos.
Como os sinais WNT viajam pelo complexo PKD1–PKD2
Para rastrear essa cadeia de sinalização, os pesquisadores usaram biossensores baseados em luz que relatam como as proteínas G se rearranjam em células vivas. Em células humanas derivadas do rim, engenheiradas para expressar PKD1, vários membros diferentes da família WNT fizeram subunidades específicas das proteínas G se separar de suas parceiras — uma marca de ativação. A resposta foi mais forte para um subtipo inibitório de proteína G, conhecido como Gαi3, e também envolveu as subunidades relacionadas Gαi1, Gαi2 e Gαq, mas não as proteínas G que tipicamente aumentam o cAMP. Biósensores adicionais mostraram que a estimulação por WNT não apenas aproximou as proteínas G do PKD1, mas também impulsionou a etapa química em que GDP é trocado por GTP na subunidade Gα, completando o ciclo de ativação. Esses efeitos ocorreram mesmo quando outros receptores WNT conhecidos foram suprimidos, indicando que o próprio PKD1, e não os receptores WNT clássicos, era o responsável.
PKD2 como parceira essencial e guardiã
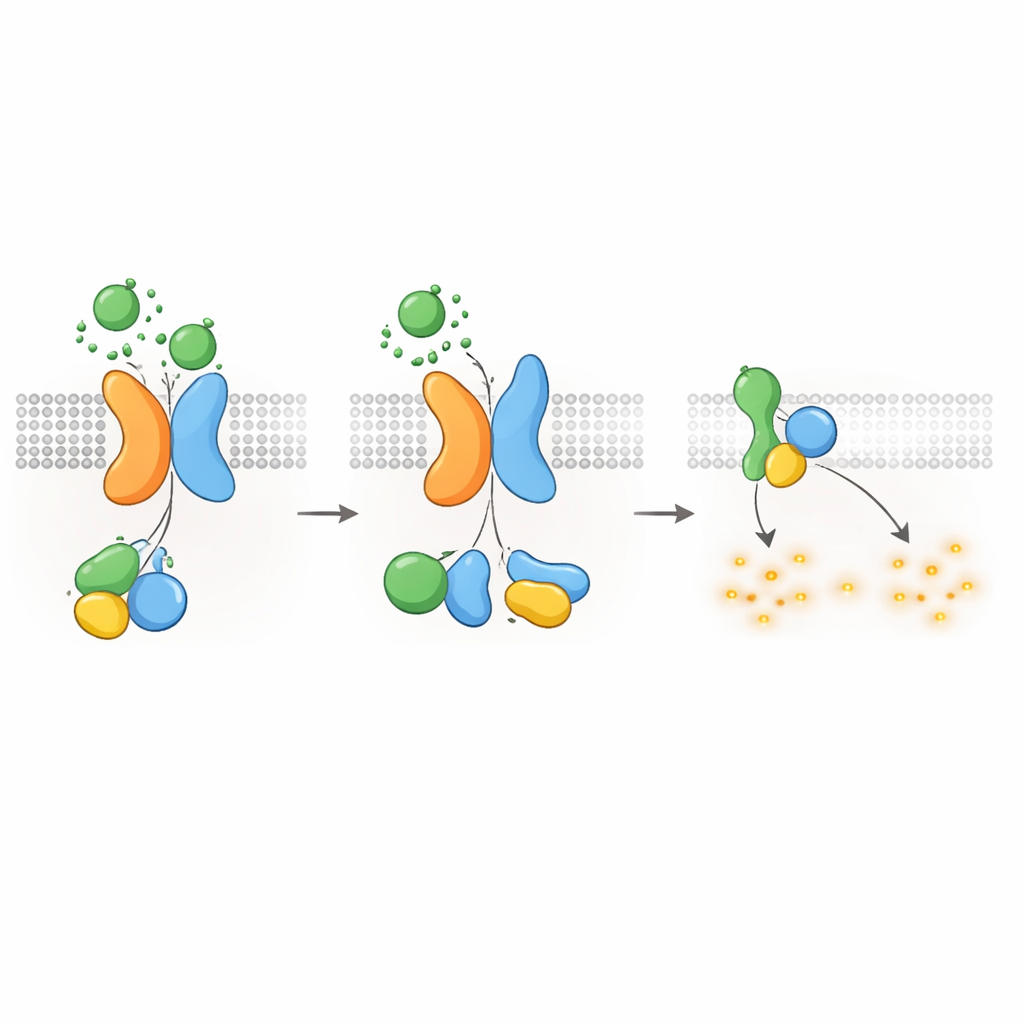
Embora o PKD1 possa se ligar tanto a ligantes WNT quanto a proteínas G, ele não atua sozinho. A equipe usou edição genômica para remover o PKD2 das células e constatou que a WNT não conseguia mais induzir a ativação de proteínas G via PKD1. A reintrodução do PKD2 normal restaurou a sinalização, mas variantes de PKD2 associadas à doença que atrapalham a montagem do complexo ou o direcionamento à membrana não o fizeram. Microscopia e medições de transferência de energia mostraram que o PKD2 atua como uma chaperona, escoltando o PKD1 até a superfície celular e estabilizando-o ali. Importante, mutações que alteram a atividade de canal iônico do PKD2 sem afetar sua parceria com o PKD1 deixaram a sinalização via proteínas G intacta, e bloquear o fluxo iônico com um inibidor químico teve pouco efeito. Isso indica que, neste contexto, o complexo PKD1–PKD2 funciona primariamente como um receptor, e não como um canal iônico atuante.
Manter o cAMP — e o crescimento dos cistos — sob controle
Como proteínas G do tipo Gαi são conhecidas por reduzir a produção de cAMP, os autores perguntaram em seguida se o receptor PKD1–PKD2 poderia diminuir diretamente os níveis de cAMP. Usando um repórter luminescente que acompanha o cAMP em tempo real, eles mostraram que a estimulação por WNT em células que expressam PKD1 retardou o aumento de cAMP desencadeado pela forskolina, um composto que eleva a produção de cAMP. As WNT também reduziram os níveis basais de cAMP, e ambos os efeitos foram bloqueados pela toxina pertussis, que desativa especificamente proteínas Gαi. Mutações causadoras de doença no PKD1 — afetando a ligação às WNT, o processamento autoproteolítico ou a interação com proteínas G — enfraqueceram severamente essa resposta de redução de cAMP. Células sem PKD2 também não exibiram supressão de cAMP dependente de WNT, a menos que o PKD2 normal fosse reintroduzido. Essas descobertas conectam os defeitos moleculares em PKD1 ou PKD2 diretamente à perda do controle do cAMP, um motor central do crescimento de cistos na DRPAD.
Desligando o sinal e internalizando receptores
Como outros GPCRs, o complexo PKD1–PKD2 precisa ser desligado e reciclado após a ativação. Os autores descobriram que uma quinase chamada GRK6, que reside na membrana plasmática, associa-se fortemente ao PKD1 quando o PKD2 está presente e ajuda a remover o complexo da superfície celular. Outra proteína, β-arrestina 2, é então recrutada ao complexo ativado e escolta tanto o PKD1 quanto o PKD2 para endossomos iniciais dentro da célula. Bloquear a atividade da GRK6 ou a função da β-arrestina reduziu essa internalização e restaurou parcialmente a sinalização por proteínas G. Essas etapas de dessensibilização sugerem que o receptor PKD1–PKD2 é rigidamente regulado, impedindo uma supressão descontrolada do cAMP em condições normais.
O que isso significa para a doença renal e além
Em conjunto, os resultados estabelecem o complexo PKD1–PKD2 como uma nova classe de receptor acoplado à proteína G — um receptor incomum com 11 transmembranas que depende de uma subunidade parceira para posicionamento adequado na superfície celular. Em rins saudáveis, sinais WNT que atuam por esse receptor ativam proteínas G inibitórias e mantêm os níveis de cAMP em uma faixa segura, ajudando a manter o diâmetro normal dos túbulos e a prevenir a formação de cistos. Quando PKD1 ou PKD2 está mutado, essa via WNT–GPCR falha, o cAMP aumenta e os cistos se expandem. Ao esclarecer essa etapa inicial do processo da doença, o estudo abre a porta para terapias que visem restaurar ou imitar a sinalização PKD1–PKD2, potencialmente oferecendo alternativas mais precisas aos medicamentos atuais que reduzem o cAMP de forma mais ampla.
Citação: Hardy, E.P., Haider, A.N., Patel, M.M. et al. A heteromeric TRP channel that functions as a WNT-activated G protein-coupled receptor. Nat Commun 17, 3233 (2026). https://doi.org/10.1038/s41467-026-69932-w
Palavras-chave: doença renal policística, PKD1 PKD2, receptor acoplado à proteína G, sinalização WNT, cAMP