Clear Sky Science · pl
Heteromerowy kanał TRP działający jako receptor sprzężony z białkiem G aktywowany przez WNT
Dlaczego białko nerkowe ma znaczenie dla każdego
Autosomalnie dominująca choroba nerek z torbielami (ADPKD) jest jedną z najczęstszych dziedzicznych chorób nerek i ważną przyczyną niewydolności nerek na całym świecie. Przez dekady naukowcy znali główne geny zaangażowane w chorobę — PKD1 i PKD2 — lecz nie wiadomo było, co ich produkty białkowe dokładnie robią w komórkach. W artykule pokazano, że te białka tworzą nietypowy receptor na powierzchni komórki, który komunikuje się bezpośrednio z klasycznymi przełącznikami sygnałowymi wewnątrz komórki. Wyjaśniając, jak ten układ normalnie pomaga utrzymać pod kontrolą kluczowy przekaźnik molekularny, praca ta daje jaśniejszy obraz powstawania torbieli w ADPKD i wskazuje nowe sposoby leczenia choroby.
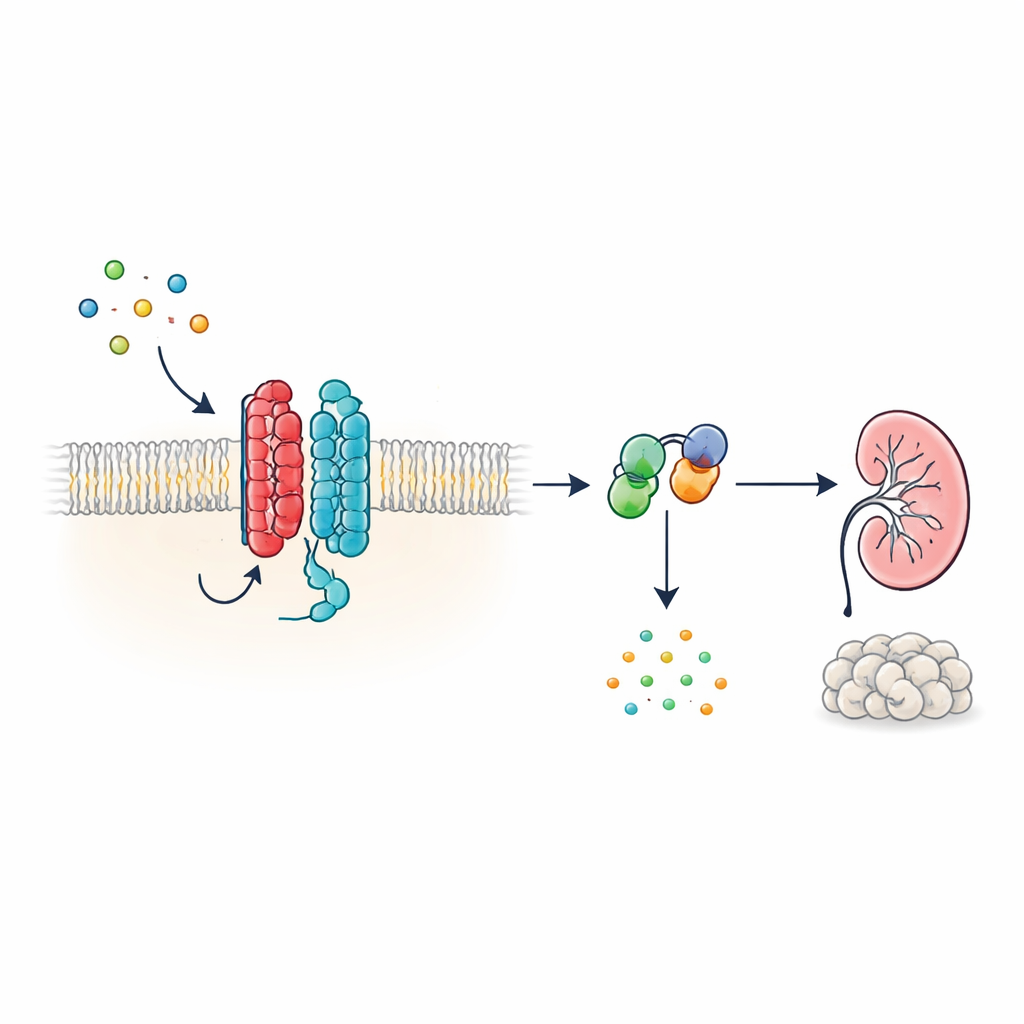
Nowy rodzaj komórkowej anteny
Wiele hormonów i sygnałów w organizmie działa za pośrednictwem receptorów sprzężonych z białkiem G (GPCR), ogromnej rodziny białek powierzchniowych o charakterystycznej strukturze siedmiu segmentów przecinających błonę komórkową. PKD1 wygląda jednak inaczej: ma 11 segmentów i była klasyfikowana bliżej kanałów jonowych niż klasycznych receptorów. Autorzy wykazują, że pomimo tej nietypowej budowy PKD1 zachowuje się jak prawdziwy GPCR, gdy współdziała z PKD2. Razem tworzą kompleks w błonie, który reaguje na białka WNT — wszechstronne molekuły sygnałowe zaangażowane w rozwój i utrzymanie tkanek. Gdy WNT wiąże się z PKD1, kompleks uruchamia wewnątrzkomórkowe białka G i zmienia stężenie cyklicznego AMP (cAMP), małej cząsteczki silnie wpływającej na wzrost komórek i wydzielanie płynów.
Jak sygnały WNT przechodzą przez kompleks PKD1–PKD2
Aby odtworzyć łańcuch sygnałowy, badacze użyli biosensorów optycznych, które wskazują, jak białka G reorganizują się w żywych komórkach. W komórkach pochodzenia nerkowego ludzkiego, zmodyfikowanych genetycznie do ekspresji PKD1, kilka członków rodziny WNT spowodowało rozdzielenie się określonych podjednostek białek G od ich partnerów — co jest znamienną cechą aktywacji. Najsilniejsza odpowiedź dotyczyła jednego podtypu hamującego białka G, znanego jako Gαi3, a także pokrewnych podjednostek Gαi1, Gαi2 i Gαq, lecz nie obejmowała białek G typowo podnoszących cAMP. Dodatkowe biosensory wykazały, że stymulacja WNT nie tylko zbliżała białka G do PKD1, lecz także napędzała krok chemiczny, w którym GDP jest wymieniane na GTP na podjednostce Gα, kończąc cykl aktywacji. Efekty te występowały nawet wtedy, gdy inne znane receptory WNT były tłumione, co wskazuje, że za odpowiedzialny jest sam PKD1, a nie klasyczne receptory WNT.
PKD2 jako niezbędny partner i strażnik
Chociaż PKD1 może wiązać zarówno ligandy WNT, jak i białka G, nie działa samodzielnie. Zespół użył edycji genomu do usunięcia PKD2 z komórek i stwierdził, że WNT nie było już w stanie uruchomić aktywacji białek G przez PKD1. Ponowne wprowadzenie normalnego PKD2 przywróciło sygnalizację, natomiast warianty PKD2 związane z chorobą, które zaburzają składanie kompleksu lub ukierunkowanie do błony, tego nie robiły. Mikroskopia i pomiary transferu energii pokazały, że PKD2 działa jak chaperon, eskortując PKD1 na powierzchnię komórki i stabilizując je tam. Co ważne, mutacje zmieniające aktywność kanałową PKD2 bez wpływu na jego partnerstwo z PKD1 pozostawiły sygnalizację przez białka G nienaruszoną, a blokowanie przepływu jonów za pomocą chemicznego inhibitora miało niewielki efekt. Wskazuje to, że w tym kontekście kompleks PKD1–PKD2 funkcjonuje głównie jako receptor, a nie pracujący kanał jonowy.
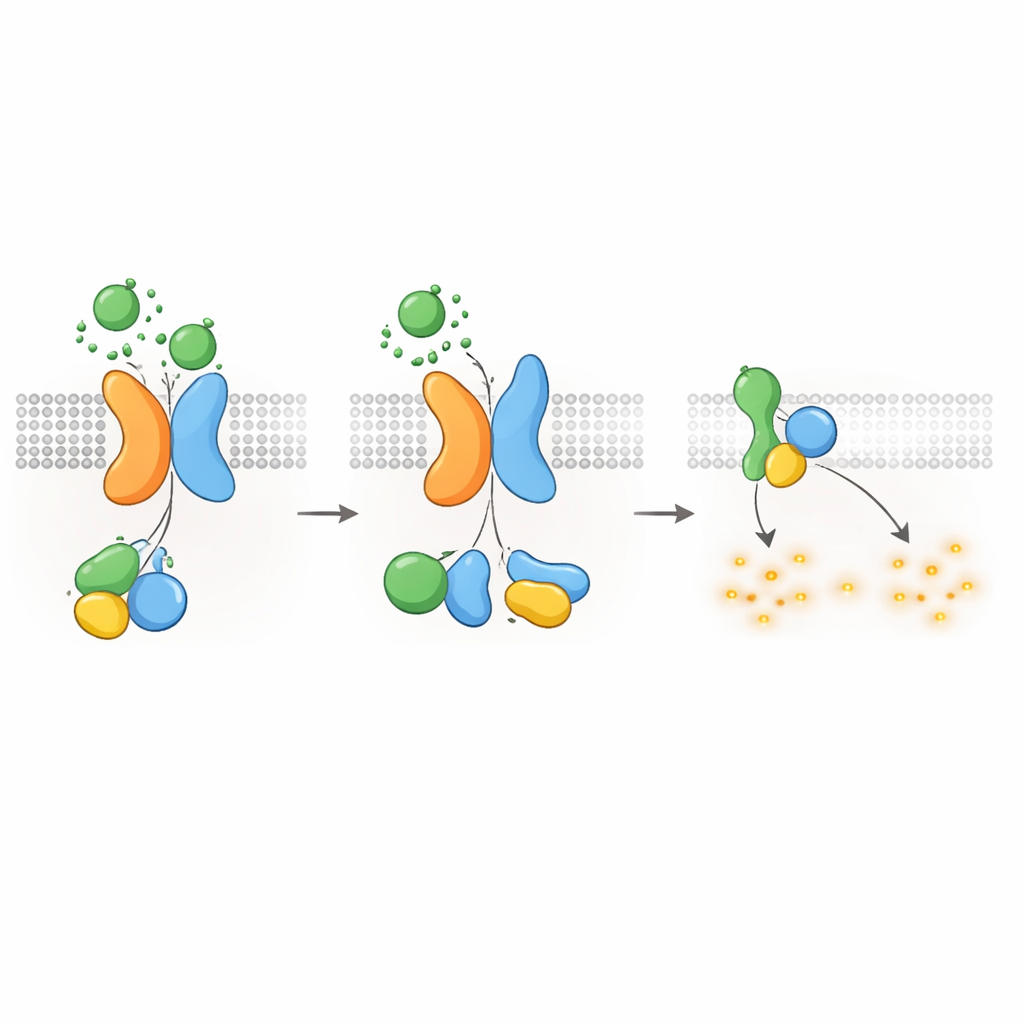
Utrzymywanie cAMP — i wzrostu torbieli — pod kontrolą
Ponieważ wiadomo, że białka G typu Gαi tłumią produkcję cAMP, autorzy zapytali następnie, czy receptor PKD1–PKD2 może bezpośrednio obniżać poziomy cAMP. Używając luminescencyjnego raportera śledzącego cAMP w czasie rzeczywistym, pokazali, że stymulacja WNT w komórkach ekspresujących PKD1 spowalniała wzrost cAMP wywołany forskoliną, związkiem zwiększającym produkcję cAMP. WNT obniżały także podstawowe poziomy cAMP, a oba efekty były blokowane przez toksynę krztuścową (pertussis), która selektywnie unieszkodliwia białka Gαi. Mutacje PKD1 powodujące chorobę — wpływające na wiązanie WNT, autoproteolityczne przetwarzanie lub interakcję z białkami G — silnie osłabiały tę odpowiedź obniżającą cAMP. Komórki pozbawione PKD2 również nie wykazywały tłumienia cAMP zależnego od WNT, chyba że ponownie wprowadzono normalne PKD2. Te wyniki łączą molekularne defekty w PKD1 lub PKD2 bezpośrednio z utratą kontroli nad cAMP, co jest kluczowym czynnikiem napędzającym wzrost torbieli w ADPKD.
Wyłączanie sygnału i przemieszczanie receptorów do wnętrza komórki
Podobnie jak inne GPCR, kompleks PKD1–PKD2 musi być wyłączony i poddany recyklingowi po aktywacji. Autorzy odkryli, że kinaza o nazwie GRK6, która znajduje się przy błonie plazmatycznej, silnie wiąże się z PKD1 w obecności PKD2 i pomaga usuwać kompleks z powierzchni komórki. Kolejne białko, β-arrestyna 2, jest wówczas rekrutowana do aktywowanego kompleksu i eskortuje zarówno PKD1, jak i PKD2 do wczesnych endosomów wewnątrz komórki. Zablokowanie aktywności GRK6 lub funkcji β-arrestyny zmniejszało tę internalizację i częściowo przywracało sygnalizację przez białka G. Te kroki desensytyzacyjne sugerują, że receptor PKD1–PKD2 jest ściśle regulowany, co zapobiega nadmiernemu tłumieniu cAMP w warunkach fizjologicznych.
Co to oznacza dla chorób nerek i nie tylko
Wyniki razem ustanawiają kompleks PKD1–PKD2 jako nową klasę receptorów sprzężonych z białkiem G — nietypowy receptor z 11 transbłonowymi segmentami, zależny od podjednostki partnerskiej dla prawidłowego umiejscowienia na powierzchni komórki. W zdrowych nerkach sygnały WNT działające przez ten receptor aktywują hamujące białka G i utrzymują poziomy cAMP w bezpiecznym zakresie, co pomaga zachować prawidłową średnicę kanalików i zapobiega tworzeniu się torbieli. Gdy PKD1 lub PKD2 jest zmutowane, droga WNT–GPCR zawodzi, cAMP rośnie, a torbiele się powiększają. Wyjaśniając ten wczesny krok w procesie chorobowym, badanie otwiera drzwi do terapii mających na celu przywrócenie lub imitowanie sygnalizacji PKD1–PKD2, co potencjalnie pozwoli na bardziej precyzyjne alternatywy wobec obecnych leków, które ogólniej tłumią cAMP.
Cytowanie: Hardy, E.P., Haider, A.N., Patel, M.M. et al. A heteromeric TRP channel that functions as a WNT-activated G protein-coupled receptor. Nat Commun 17, 3233 (2026). https://doi.org/10.1038/s41467-026-69932-w
Słowa kluczowe: choroba nerek z torbielami o podłożu genetycznym, PKD1 PKD2, receptor sprzężony z białkiem G, sygnalizacja WNT, cAMP