Clear Sky Science · de
Ein heteromerer TRP-Kanal, der als WNT-aktivierter G-Protein-gekoppelter Rezeptor fungiert
Warum ein Nierenprotein für alle wichtig ist
Die autosomal-dominante polyzystische Nierenerkrankung (ADPKD) gehört zu den häufigsten erblichen Nierenerkrankungen und ist weltweit eine wichtige Ursache für Nierenversagen. Jahrzehntelang waren die Hauptgene—PKD1 und PKD2—bekannt, doch blieb unklar, welche Funktionen ihre Proteinprodukte in der Zelle tatsächlich ausüben. Diese Studie zeigt, dass diese Proteine an der Zelloberfläche einen ungewöhnlichen Rezeptor bilden, der direkt mit klassischen intrazellulären Schaltsystemen kommuniziert. Indem sie erklärt, wie dieses System normalerweise ein zentrales Botenmolekül in Schach hält, liefert die Arbeit ein klareres Bild davon, wie Zysten bei ADPKD entstehen, und weist auf neue therapeutische Ansätze hin.
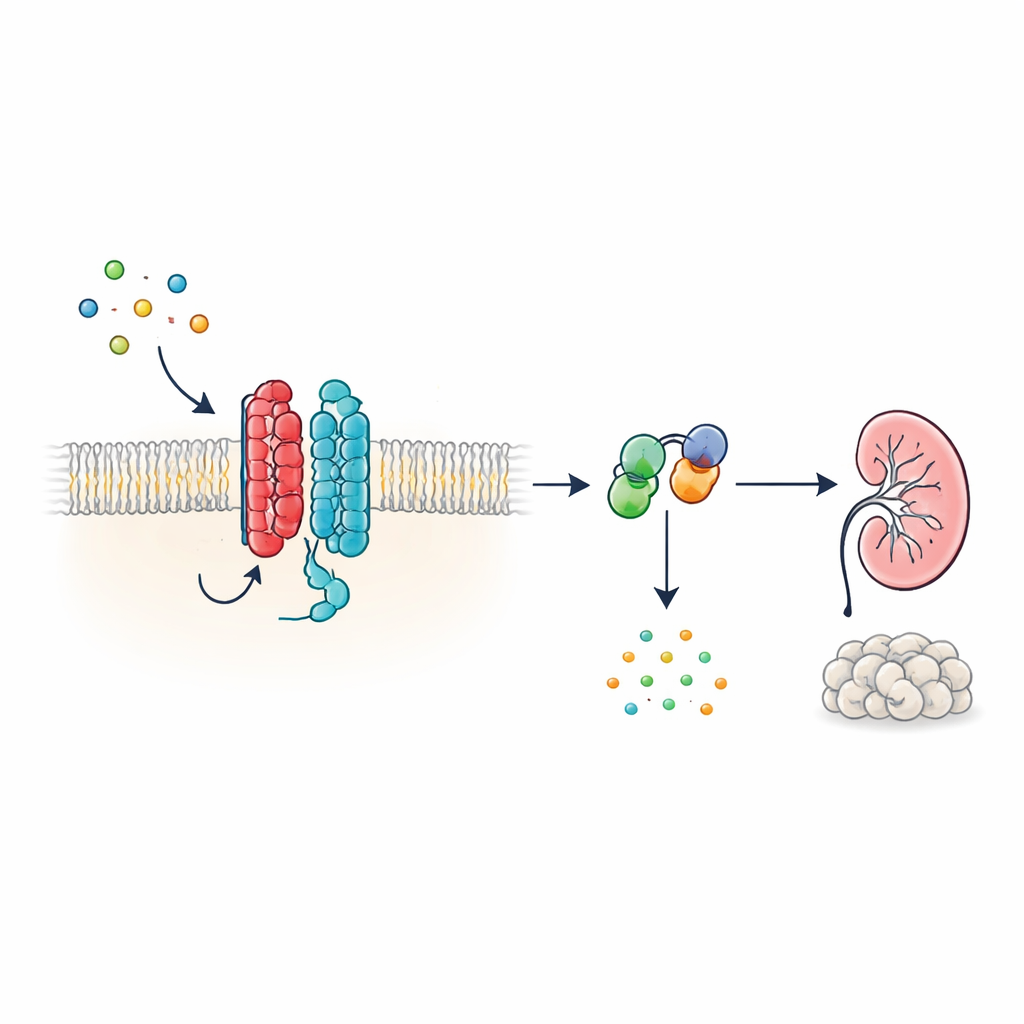
Eine neue Art zellulärer Antenne
Viele Hormone und Signale im Körper wirken über G-Protein-gekoppelte Rezeptoren (GPCRs), eine große Familie von Zelloberflächenproteinen mit einer typischen siebenfach durch die Membran verlaufenden Struktur. PKD1 sieht hingegen anders aus: Es besitzt 11 Transmembrandomänen und wurde eher mit Ionenkanälen als mit klassischen Rezeptoren in Verbindung gebracht. Die Autorinnen und Autoren zeigen, dass PKD1 trotz dieser unkonventionellen Form als echter GPCR agiert, wenn es mit PKD2 gepaart ist. Gemeinsam bilden die beiden Proteine einen Komplex in der Membran, der auf WNT-Proteine reagiert—vielseitige Signalmoleküle, die an Entwicklung und Gewebehomöostase beteiligt sind. Binden WNTs an PKD1, löst der Komplex intrazelluläre G-Proteine aus und verändert die Konzentration von cyclischem AMP (cAMP), einem kleinen Molekül, das Zellwachstum und Flüssigkeitssekretion stark beeinflusst.
Wie WNT-Signale durch den PKD1–PKD2-Komplex reisen
Um diese Signalkette nachzuverfolgen, setzten die Forscher lichtbasierte Biosensoren ein, die anzeigen, wie sich G-Proteine in lebenden Zellen umordnen. In humanen, aus Nieren stammenden Zellen, die so verändert wurden, dass sie PKD1 exprimieren, lösten mehrere verschiedene WNT-Familienmitglieder ein spezifisches Auseinanderfallen von G-Protein-Untereinheiten aus—ein Kennzeichen der Aktivierung. Die stärkste Antwort zeigte sich bei einem inhibitorischen G-Protein-Subtype, bekannt als Gαi3; auch verwandte Untereinheiten Gαi1, Gαi2 und Gαq waren beteiligt, nicht jedoch die G-Proteine, die typischerweise cAMP erhöhen. Weitere Biosensoren zeigten, dass WNT-Stimulation nicht nur G-Proteine in die Nähe von PKD1 brachte, sondern auch den chemischen Schritt förderte, bei dem GDP auf der Gα-Untereinheit gegen GTP ausgetauscht wird und damit der Aktivierungszyklus vollendet wird. Diese Effekte traten auch auf, wenn andere bekannte WNT-Rezeptoren unterdrückt waren, was darauf hindeutet, dass PKD1 selbst—und nicht klassische WNT-Rezeptoren—verantwortlich ist.
PKD2 als unverzichtbarer Partner und Torwächter
Obwohl PKD1 sowohl WNT-Liganden als auch G-Proteine binden kann, agiert es nicht allein. Das Team entfernte PKD2 mittels Genomeditierung aus Zellen und fand heraus, dass WNT dann PKD1 nicht mehr zur Aktivierung von G-Proteinen bringen konnte. Die Wiedereinführung normalen PKD2 stellte die Signalübertragung wieder her, krankheitsassoziierte PKD2-Varianten, die die Komplexbildung oder Membranlokalisierung stören, taten dies jedoch nicht. Mikroskopie und Energieübertragungsmessungen zeigten, dass PKD2 als Chaperon wirkt, PKD1 zur Zelloberfläche begleitet und dort stabilisiert. Wichtig ist, dass Mutationen, die die Ionenkanalaktivität von PKD2 verändern, ohne die Partnerschaft mit PKD1 zu beeinflussen, die G-Protein-Signalübertragung nicht beeinträchtigen; und das Blockieren des Ionenflusses mit einem chemischen Inhibitor hatte kaum Wirkung. Dies deutet darauf hin, dass der PKD1–PKD2-Komplex in diesem Zusammenhang vorwiegend als Rezeptor und weniger als funktionaler Ionenkanal fungiert.
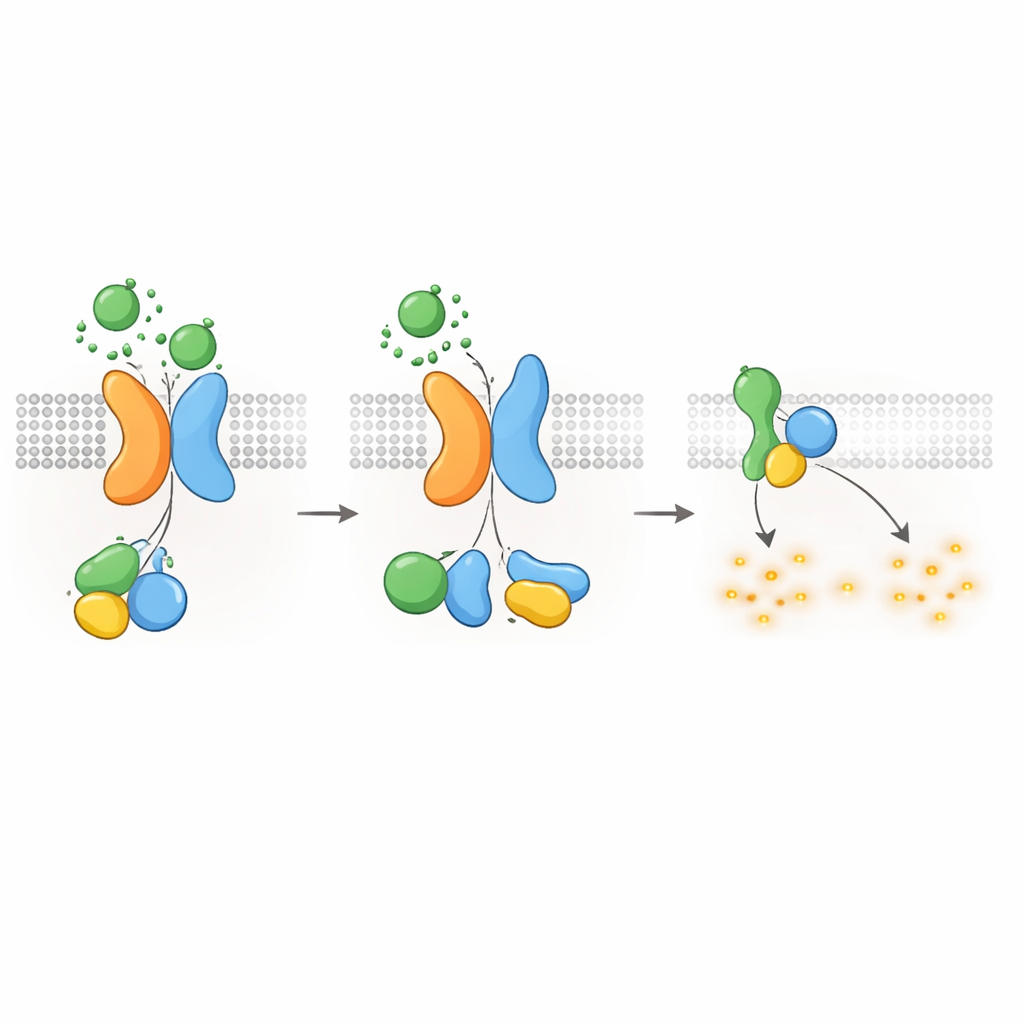
cAMP — und Zystenwachstum — in Schach halten
Da Gαi-ähnliche G-Proteine dafür bekannt sind, die cAMP-Produktion zu dämpfen, fragten die Autorinnen und Autoren als Nächstes, ob der PKD1–PKD2-Rezeptor cAMP-Spiegel direkt senken kann. Mithilfe eines lumineszenten Reporters, der cAMP in Echtzeit verfolgt, zeigten sie, dass WNT-Stimulation in PKD1-exprimierenden Zellen den Anstieg von cAMP, der durch Forskolin ausgelöst wurde, verlangsamte; Forskolin ist eine Verbindung, die die cAMP-Produktion erhöht. WNTs senkten auch die basalen cAMP-Spiegel, und beide Effekte wurden durch Pertussis-Toxin blockiert, das spezifisch Gαi-Proteine deaktiviert. Krankheitsverursachende PKD1-Mutationen—die WNT-Bindung, autoproteolytische Verarbeitung oder G-Protein-Interaktion betreffen—schwächten diese cAMP-senkende Antwort stark ab. Zellen ohne PKD2 zeigten ebenfalls keine WNT-abhängige cAMP-Unterdrückung, sofern nicht normales PKD2 wieder eingeführt wurde. Diese Befunde verknüpfen die molekularen Defekte in PKD1 oder PKD2 direkt mit dem Verlust der cAMP-Kontrolle, einem zentralen Treiber des Zystenwachstums bei ADPKD.
Signalabschaltung und Rezeptorinternealisierung
Wie andere GPCRs muss auch der PKD1–PKD2-Komplex nach der Aktivierung abgeschaltet und recycelt werden. Die Autoren fanden, dass eine Kinase namens GRK6, die an der Plasmamembran lokalisiert ist, stark mit PKD1 assoziiert, wenn PKD2 vorhanden ist, und dabei hilft, den Komplex von der Zelloberfläche zu entfernen. Ein weiteres Protein, β-Arrestin 2, wird dann an den aktivierten Komplex rekrutiert und begleitet sowohl PKD1 als auch PKD2 in frühe Endosomen innerhalb der Zelle. Das Blockieren der GRK6-Aktivität oder der β-Arrestin-Funktion verringerte diese Internalisierung und stellte teilweise die G-Protein-Signalübertragung wieder her. Diese Desensibilisierungsschritte deuten darauf hin, dass der PKD1–PKD2-Rezeptor streng reguliert ist, um eine übermäßige Unterdrückung von cAMP unter normalen Bedingungen zu verhindern.
Was das für Nierenerkrankungen und darüber hinaus bedeutet
In der Summe stellen die Ergebnisse den PKD1–PKD2-Komplex als eine neue Klasse von G-Protein-gekoppelten Rezeptoren dar—einen unkonventionellen 11-Transmembran-Rezeptor, der für die korrekte Platzierung an der Zelloberfläche von einer Partneruntereinheit abhängt. In gesunden Nieren aktivieren WNT-Signale über diesen Rezeptor inhibitorische G-Proteine und halten die cAMP-Spiegel in einem sicheren Bereich, was hilft, den normalen Tubulusdurchmesser zu erhalten und Zystenbildung zu verhindern. Sind PKD1 oder PKD2 mutiert, versagt dieser WNT–GPCR-Weg, cAMP steigt und Zysten vergrößern sich. Indem die Studie diesen frühen Schritt im Krankheitsprozess klärt, eröffnet sie Wege für Therapien, die darauf abzielen, PKD1–PKD2-Signale wiederherzustellen oder zu imitieren, und bietet damit potenziell präzisere Alternativen zu aktuellen Medikamenten, die cAMP breiter dämpfen.
Zitation: Hardy, E.P., Haider, A.N., Patel, M.M. et al. A heteromeric TRP channel that functions as a WNT-activated G protein-coupled receptor. Nat Commun 17, 3233 (2026). https://doi.org/10.1038/s41467-026-69932-w
Schlüsselwörter: polyzystische Nierenerkrankung, PKD1 PKD2, G-Protein-gekoppelter Rezeptor, WNT-Signalübertragung, cAMP