Clear Sky Science · en
A heteromeric TRP channel that functions as a WNT-activated G protein-coupled receptor
Why a kidney protein matters to everyone
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common inherited kidney disorders and a major cause of kidney failure worldwide. For decades, scientists have known the main genes involved—PKD1 and PKD2—but not what their protein products actually do inside cells. This paper reveals that these proteins form an unusual receptor at the cell surface that talks directly to classic signaling switches inside the cell. By explaining how this system normally helps keep a key messenger molecule in check, the work offers a clearer picture of how cysts arise in ADPKD and points to new ways to treat the disease.
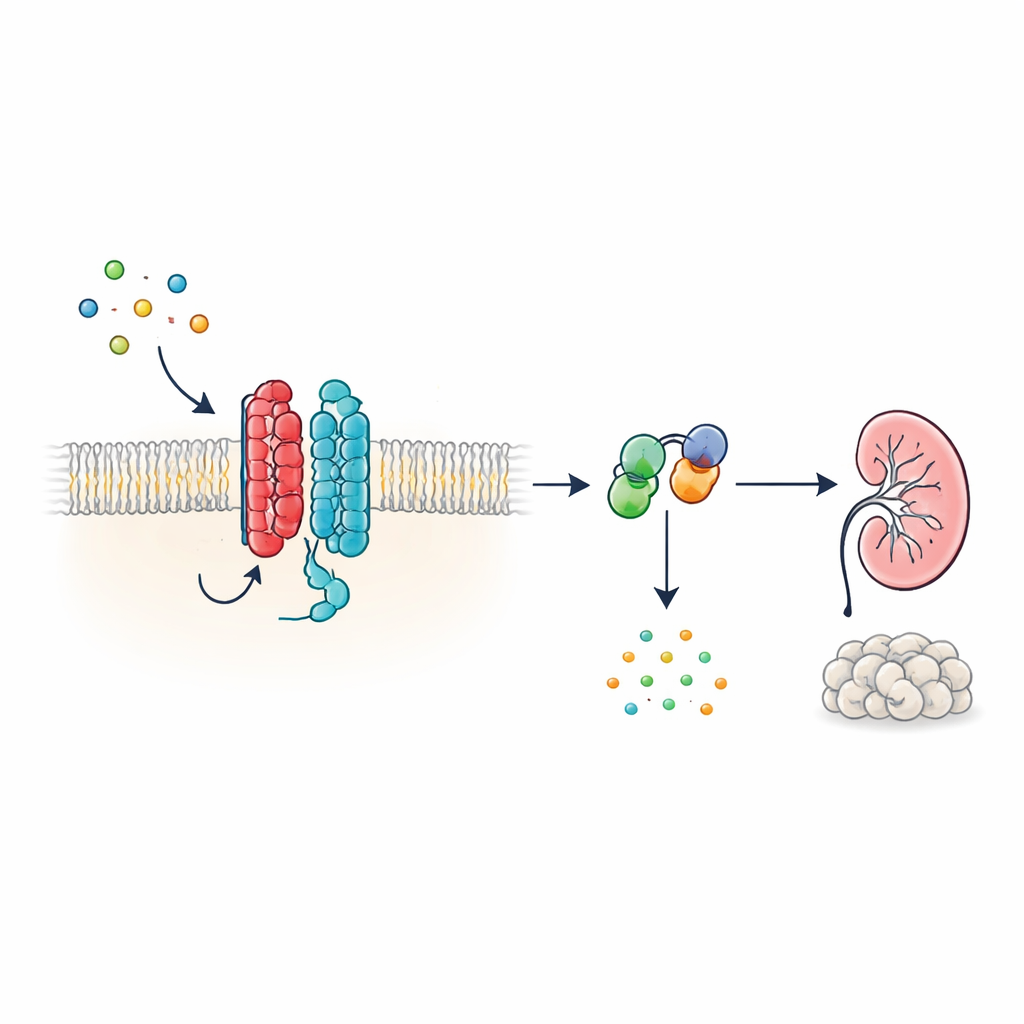
A new kind of cellular antenna
Many hormones and signals in the body act through G protein-coupled receptors (GPCRs), a huge family of cell-surface proteins with a characteristic seven-segment structure that threads through the cell membrane. PKD1, however, looks different: it has 11 segments and has been grouped with ion channels rather than classic receptors. The authors show that, despite this unconventional shape, PKD1 behaves as a bona fide GPCR when paired with PKD2. Together, the two proteins form a complex in the membrane that responds to WNT proteins—versatile signaling molecules involved in development and tissue maintenance. When WNTs bind PKD1, the complex triggers intracellular G proteins and alters levels of cyclic AMP (cAMP), a small molecule that strongly influences cell growth and fluid secretion.
How WNT signals travel through the PKD1–PKD2 complex
To trace this signaling chain, the researchers used light-based biosensors that report how G proteins rearrange in living cells. In human kidney-derived cells engineered to express PKD1, several different WNT family members caused specific G protein subunits to split apart from their partners—a hallmark of activation. The response was strongest for one inhibitory G protein subtype, known as Gαi3, and also involved related Gαi1, Gαi2 and Gαq subunits, but not the G proteins that typically raise cAMP. Additional biosensors showed that WNT stimulation not only brought G proteins into close proximity with PKD1, but also drove the chemical step in which GDP is swapped for GTP on the Gα subunit, completing the activation cycle. These effects occurred even when other known WNT receptors were suppressed, indicating that PKD1 itself, rather than classical WNT receptors, was responsible.
PKD2 as the essential partner and gatekeeper
Although PKD1 can bind both WNT ligands and G proteins, it does not act alone. The team used genome editing to remove PKD2 from cells and found that WNT could no longer drive G protein activation through PKD1. Reintroducing normal PKD2 restored signaling, but disease-associated PKD2 variants that disrupt complex assembly or membrane targeting did not. Microscopy and energy-transfer measurements showed that PKD2 acts as a chaperone, escorting PKD1 to the cell surface and stabilizing it there. Importantly, mutations that change PKD2’s ion-channel activity without affecting its partnership with PKD1 left G protein signaling intact, and blocking ion flow with a chemical inhibitor had little effect. This indicates that, in this context, the PKD1–PKD2 complex functions primarily as a receptor rather than as a working ion channel.
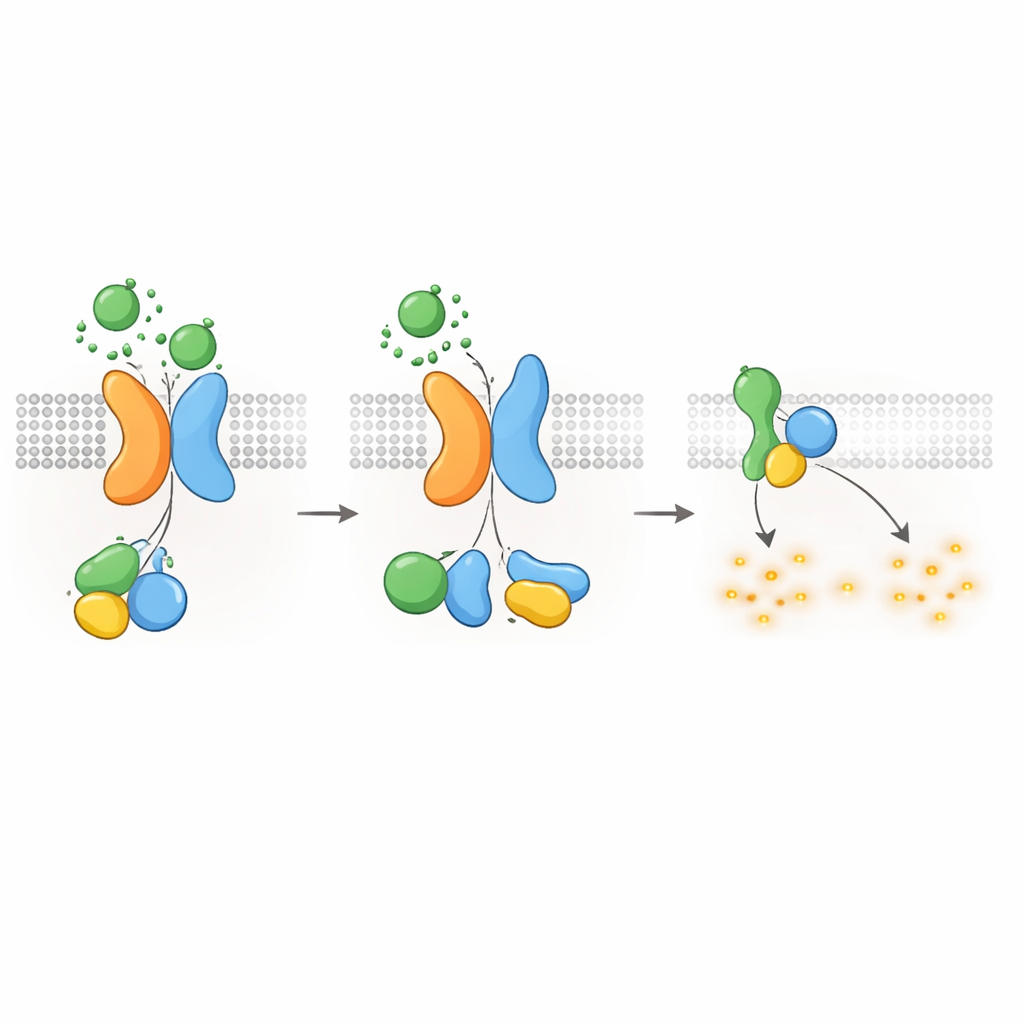
Keeping cAMP—and cyst growth—under control
Since Gαi-type G proteins are known to dampen cAMP production, the authors next asked whether the PKD1–PKD2 receptor could directly lower cAMP levels. Using a luminescent reporter that tracks cAMP in real time, they showed that WNT stimulation in PKD1-expressing cells slowed the rise of cAMP triggered by forskolin, a compound that boosts cAMP production. WNTs also lowered resting cAMP levels, and both effects were blocked by pertussis toxin, which specifically disables Gαi proteins. Disease-causing PKD1 mutations—affecting WNT binding, autoproteolytic processing, or G protein interaction—severely weakened this cAMP-lowering response. Cells lacking PKD2 also failed to show WNT-dependent cAMP suppression, unless normal PKD2 was reintroduced. These findings connect the molecular defects in PKD1 or PKD2 directly to loss of cAMP restraint, a central driver of cyst growth in ADPKD.
Switching off the signal and moving receptors inside
Like other GPCRs, the PKD1–PKD2 complex must be turned off and recycled after activation. The authors found that a kinase called GRK6, which resides at the plasma membrane, associates strongly with PKD1 when PKD2 is present and helps remove the complex from the cell surface. Another protein, β-arrestin 2, is then recruited to the activated complex and escorts both PKD1 and PKD2 into early endosomes inside the cell. Blocking GRK6 activity or β-arrestin function reduced this internalization and partially restored G protein signaling. These desensitization steps suggest that the PKD1–PKD2 receptor is tightly regulated, preventing runaway suppression of cAMP under normal conditions.
What this means for kidney disease and beyond
Together, the results establish the PKD1–PKD2 complex as a new class of G protein-coupled receptor—an unconventional 11-transmembrane receptor that depends on a partner subunit for proper placement at the cell surface. In healthy kidneys, WNT signals acting through this receptor activate inhibitory G proteins and keep cAMP levels in a safe range, helping to maintain normal tubule diameter and prevent cyst formation. When PKD1 or PKD2 is mutated, this WNT–GPCR pathway falters, cAMP rises, and cysts expand. By clarifying this early step in the disease process, the study opens the door to therapies aimed at restoring or mimicking PKD1–PKD2 signaling, potentially offering more precise alternatives to current drugs that blunt cAMP more broadly.
Citation: Hardy, E.P., Haider, A.N., Patel, M.M. et al. A heteromeric TRP channel that functions as a WNT-activated G protein-coupled receptor. Nat Commun 17, 3233 (2026). https://doi.org/10.1038/s41467-026-69932-w
Keywords: polycystic kidney disease, PKD1 PKD2, G protein-coupled receptor, WNT signaling, cAMP