Clear Sky Science · ar
قناة TRP مغايرة التركيب تعمل كمستقبل مرتبط ببروتين G يُفعل بواسطة WNT
لماذا يهم بروتين الكلى الجميع
مرض الكلى الكلوي المتعدد الكيسات المهيمن ذاتياً (ADPKD) هو واحد من أكثر الاضطرابات الكلوية الوراثية شيوعًا وسبب رئيسي لفشل الكلى على مستوى العالم. لعقود، عرف العلماء الجينات الرئيسية المتورطة—PKD1 وPKD2—لكنهم لم يعرفوا ما الذي تفعله نواتج هذه الجينات داخل الخلايا فعلاً. تكشف هذه الورقة أن هذه البروتينات تُشكّل مستقبلًا غير عادي على سطح الخلية يتواصل مباشرة مع مفاتيح الإشارة الكلاسيكية داخل الخلية. من خلال توضيح كيف يساعد هذا النظام عادةً في ضبط رسول خلوية رئيسي، تقدم الدراسة صورة أوضح لكيفية نشوء الأكياس في ADPKD وتشير إلى طرق جديدة لعلاج المرض.
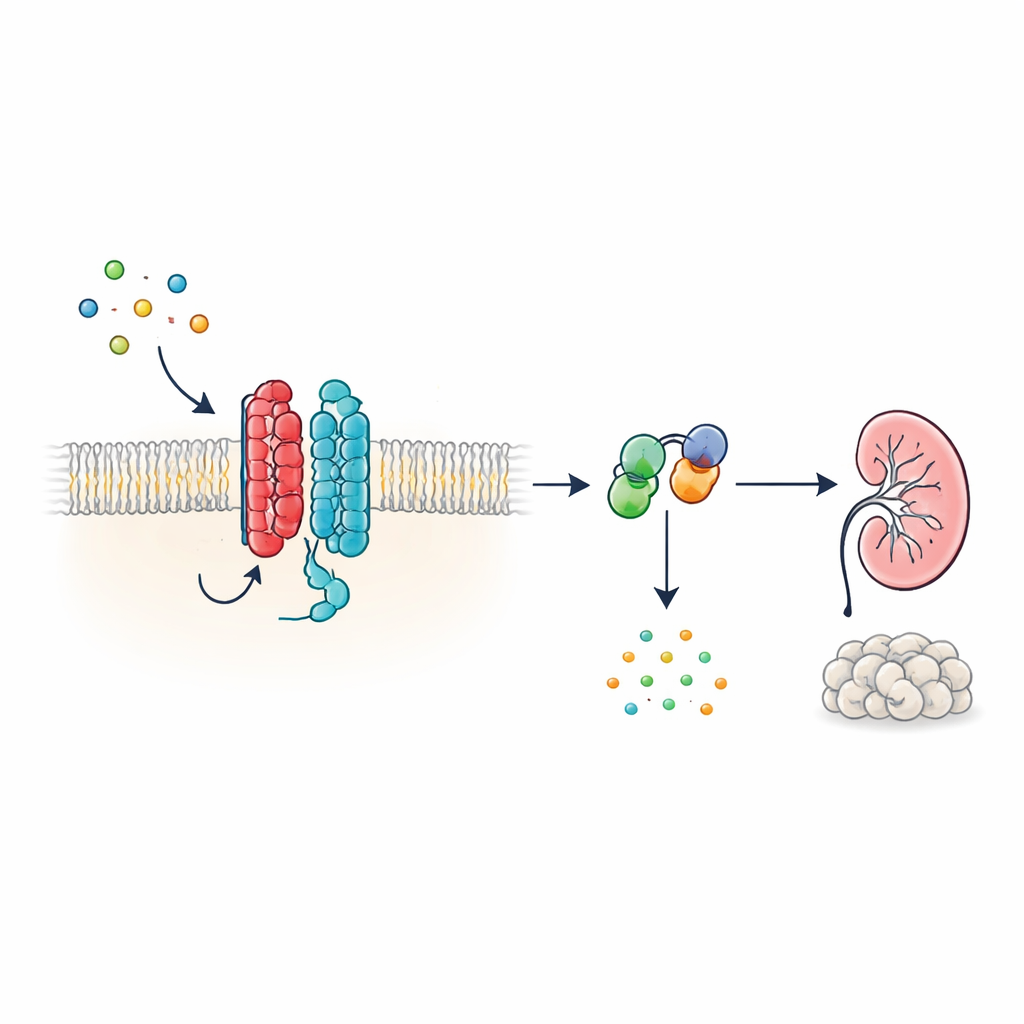
هوائي خلوي من نوع جديد
العديد من الهرمونات والإشارات في الجسم تعمل عبر مستقبلات مرتبطة ببروتين G (GPCRs)، وهي عائلة ضخمة من بروتينات سطح الخلية ذات بنية مميزة مكوّنة من سبعة مقاطع تمر عبر غشاء الخلية. يبدو PKD1، مع ذلك، مختلفًا: يحتوي على 11 مقطعًا وتم تصنيفه مع قنوات الأيونات بدلًا من المستقبلات الكلاسيكية. يظهر المؤلفون أن PKD1، بالرغم من هذا الشكل غير الاعتيادي، يتصرف كمستقبل مرتبط ببروتين G حقيقي عند اقترانه بـ PKD2. معًا، يشكل البروتينان مركبًا في الغشاء يستجيب لبروتينات WNT—جزيئات إشارات متعددة الاستخدامات مشاركة في التطور وصيانة الأنسجة. عندما ترتبط WNTs بـ PKD1، يحفّز المركب بروتينات G داخل الخلية ويغير مستويات الأدينوزين أحادي الحلقي (cAMP)، وهو جزيء صغير يؤثر بشدة على نمو الخلايا وإفراز السوائل.
كيف تنتقل إشارات WNT عبر مركب PKD1–PKD2
لتتبع سلسلة الإشارة هذه، استخدم الباحثون مستشعرات حيوية ضوئية تُبلغ كيف تعيد بروتينات G ترتيب نفسها في الخلايا الحية. في خلايا مشتقة من الكلى البشرية تم تعديلها لتعبّر عن PKD1، تسببت عدة أعضاء من عائلة WNT في فصل وحدات فرعية محددة من بروتينات G عن شركائها—وهو علامة مميزة للتنشيط. كانت الاستجابة الأقوى لنوع واحد مثبط من بروتينات G، المعروف باسم Gαi3، وشملت أيضًا الوحدات ذات الصلة Gαi1 وGαi2 وGαq، لكن ليس بروتينات G التي عادةً ترفع مستوى cAMP. أظهرت مستشعرات إضافية أن تحفيز WNT لم يجلب بروتينات G فقط إلى قرب PKD1، بل دفع أيضًا الخطوة الكيميائية التي يتم فيها استبدال GDP بـ GTP على الوحدة الفرعية Gα، مكملاً دورة التنشيط. حدثت هذه التأثيرات حتى عندما تم قمع مستقبلات WNT المعروفة الأخرى، مما يشير إلى أن PKD1 نفسه، وليس مستقبلات WNT الكلاسيكية، كان مسؤولًا.
PKD2 كشريك أساسي وحارس للبوابة
على الرغم من أن PKD1 يمكنه ربط ليجندات WNT وبروتينات G، إلا أنه لا يعمل بمفرده. استخدمت الفريق تقنيات تحرير الجينوم لإزالة PKD2 من الخلايا ووجدوا أن WNT لم يعد قادرًا على دفع تنشيط بروتينات G عبر PKD1. أعاد إدخال PKD2 الطبيعي الإشارة، لكن متغيرات PKD2 المرتبطة بالمرض والتي تعطل تجميع المركب أو استهدافه للغشاء لم تفعل ذلك. أظهرت الميكروسكوبية وقياسات نقل الطاقة أن PKD2 يعمل كمرشد، يصحب PKD1 إلى سطح الخلية ويثبته هناك. والأهم، أن الطفرات التي تغير نشاط قناة الأيونات في PKD2 دون أن تؤثر على شراكته مع PKD1 تركت إشارات بروتين G سليمة، وإيقاف تدفق الأيونات بمثبط كيميائي كان له أثر ضئيل. هذا يشير إلى أنه، في هذا السياق، يعمل مركب PKD1–PKD2 أساسًا كمستقبل بدلًا من كونه قناة أيونات فعّالة.
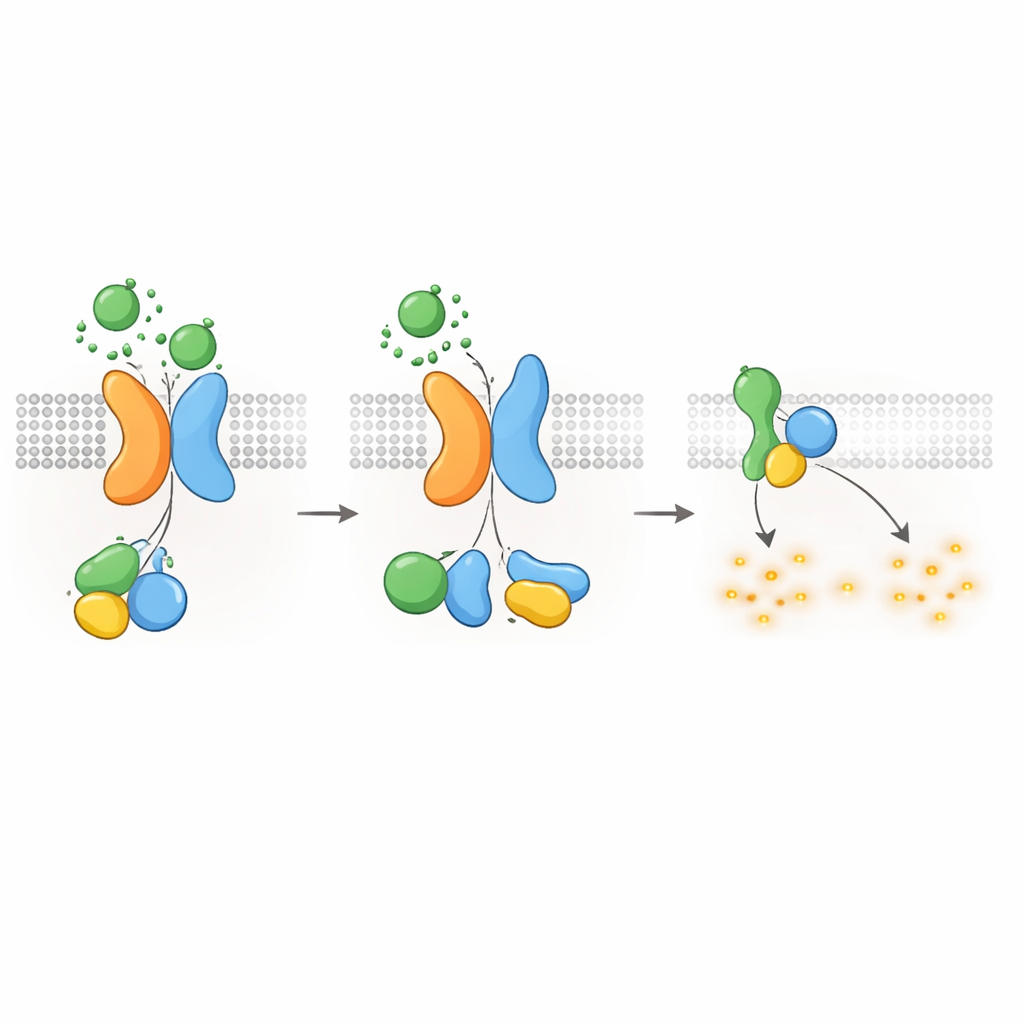
حفظ cAMP—ونمو الأكياس—تحت السيطرة
بما أن بروتينات G من نوع Gαi معروفة بتقليل إنتاج cAMP، سأل المؤلفون بعد ذلك ما إذا كان مستقبل PKD1–PKD2 يمكنه تقليل مستويات cAMP مباشرة. باستخدام مراسل مضيء يتتبع cAMP في الوقت الحقيقي، أظهروا أن تحفيز WNT في خلايا معبّرة عن PKD1 أبطأ ارتفاع cAMP الناتج عن الفورسكولين، المركب الذي يعزز إنتاج cAMP. كما خفضت WNTs مستويات cAMP الراهنة، وكلا التأثيرين تم حجبهما بواسطة سم الخناق (pertussis toxin)، الذي يعطل بروتينات Gαi بشكل محدد. أضعفت طفرات PKD1 المسببة للمرض—التي تؤثر على ربط WNT أو المعالجة الذاتية البروتوليتية أو تفاعل بروتين G—بشدة هذه الاستجابة الخافضة لـ cAMP. كما فشلت الخلايا الخالية من PKD2 في إظهار كبح cAMP المعتمد على WNT، ما لم يُعاد إدخال PKD2 الطبيعي. تربط هذه النتائج العيوب الجزيئية في PKD1 أو PKD2 مباشرة بفقدان تقييد cAMP، وهو محرك مركزي لنمو الأكياس في ADPKD.
إيقاف الإشارة وتحريك المستقبلات إلى الداخل
مثل غيرها من مستقبلات GPCR، يجب إيقاف مركب PKD1–PKD2 وإعادة تدويره بعد التنشيط. وجد المؤلفون أن كينازًا يسمى GRK6، الذي يتواجد عند غشاء البلازما، يرتبط بقوة بـ PKD1 عندما يكون PKD2 حاضرًا ويساعد في إزالة المركب من سطح الخلية. ثم يُستدعى بروتين آخر، β-arrestin 2، إلى المركب المنشط ويرافق كلًا من PKD1 وPKD2 إلى الحويصلات المبكرة داخل الخلية. أدى حجب نشاط GRK6 أو وظيفة β-arrestin إلى تقليل هذا الإدخال إلى داخل الخلية واستعادة جزئية لإشارات بروتين G. تشير هذه الخطوات في إزالة التحفيز إلى أن مستقبل PKD1–PKD2 يخضع لتنظيم محكم، مما يمنع كبحًا مفرطًا لـ cAMP في الظروف العادية.
ماذا يعني هذا لمرض الكلى وما يتجاوزها
تثبت النتائج معًا أن مركب PKD1–PKD2 يشكل فئة جديدة من المستقبلات المرتبطة ببروتين G—مستقبل غير تقليدي ذا 11 مقطعًا عبر الغشاء يعتمد على وحدة شريكة لوضعه الصحيح على سطح الخلية. في الكلى السليمة، تؤدي إشارات WNT عبر هذا المستقبل إلى تنشيط بروتينات G المثبطة والحفاظ على مستويات cAMP في نطاق آمن، مما يساعد على الحفاظ على قطر النبيبات الطبيعي ومنع تكوّن الأكياس. عندما يتعرض PKD1 أو PKD2 لطفرة، يضعف هذا المسار WNT–GPCR، يرتفع cAMP، وتتوسع الأكياس. من خلال توضيح هذه الخطوة المبكرة في مسار المرض، تفتح الدراسة الباب أمام علاجات تهدف إلى استعادة أو محاكاة إشارات PKD1–PKD2، مما قد يوفر بدائل أكثر دقة للأدوية الحالية التي تخفض cAMP على نحو أوسع.
الاستشهاد: Hardy, E.P., Haider, A.N., Patel, M.M. et al. A heteromeric TRP channel that functions as a WNT-activated G protein-coupled receptor. Nat Commun 17, 3233 (2026). https://doi.org/10.1038/s41467-026-69932-w
الكلمات المفتاحية: مرض الكلى الكلوي المتعدد الكيسات الوراثي, PKD1 PKD2, مستقبل مرتبط ببروتين G, إشارة WNT, cAMP