Clear Sky Science · pl
Korekcja molekularnego fenotypu dystonii parkinsonowskiej sprzężonej z chromosomem X ujawnia niekanoniczną funkcję BRD4
Dlaczego ta historia o chorobie mózgu ma znaczenie
Dystonia‑Parkinsonizm sprzężony z chromosomem X (XDP) to wyniszczające zaburzenie ruchowe, które dotyka przede wszystkim mężczyzn z Filipin, powodując skrętne ruchy i sztywność podobną do parkinsonizmu, nasilające się z czasem. Chociaż naukowcy znają mutację genetyczną wywołującą XDP, nie rozumieli dokładnie, w jaki sposób ta zmiana zakłóca przekazy genetyczne w komórkach nerwowych — ani jak to naprawić. To badanie odkrywa ukrytą słabość w sposobie, w jaki komórki mózgowe kopiują i kończą komunikaty genetyczne, i pokazuje, że osłabienie działania znanego białka regulatorowego BRD4 może przywrócić bardziej normalne transkrypty. Praca ta otwiera nowe spojrzenie na terapię: zamiast jedynie zastępować brakujące białko, można próbować naprawić sam proces przetwarzania komunikatu.
Dziwny insert genetyczny, który wytrąca z równowagi kluczowy gen mózgowy
W każdej komórce geny są nieustannie przepisywane na cząsteczki mRNA, które kierują produkcją białek. W XDP odcinek ruchomego DNA zwany elementem SVA wstawił się do dużego genu o nazwie TAF1, który pomaga inicjować odczyt tysięcy innych genów. Ten insert znajduje się głęboko w jednej z długich, niekodujących części TAF1. Komórki pacjentów wytwarzają około 20% mniej pełnej długości transkryptu TAF1, co zaburza sieci genów niezbędnych dla zdrowych neuronów. Nie było jasne, w jaki sposób ta obca sekwencja przerywa proces przepisywania i czy da się namówić komórki, aby ominęły ten blok.
Budowa żywego sensora wadliwych komunikatów genowych
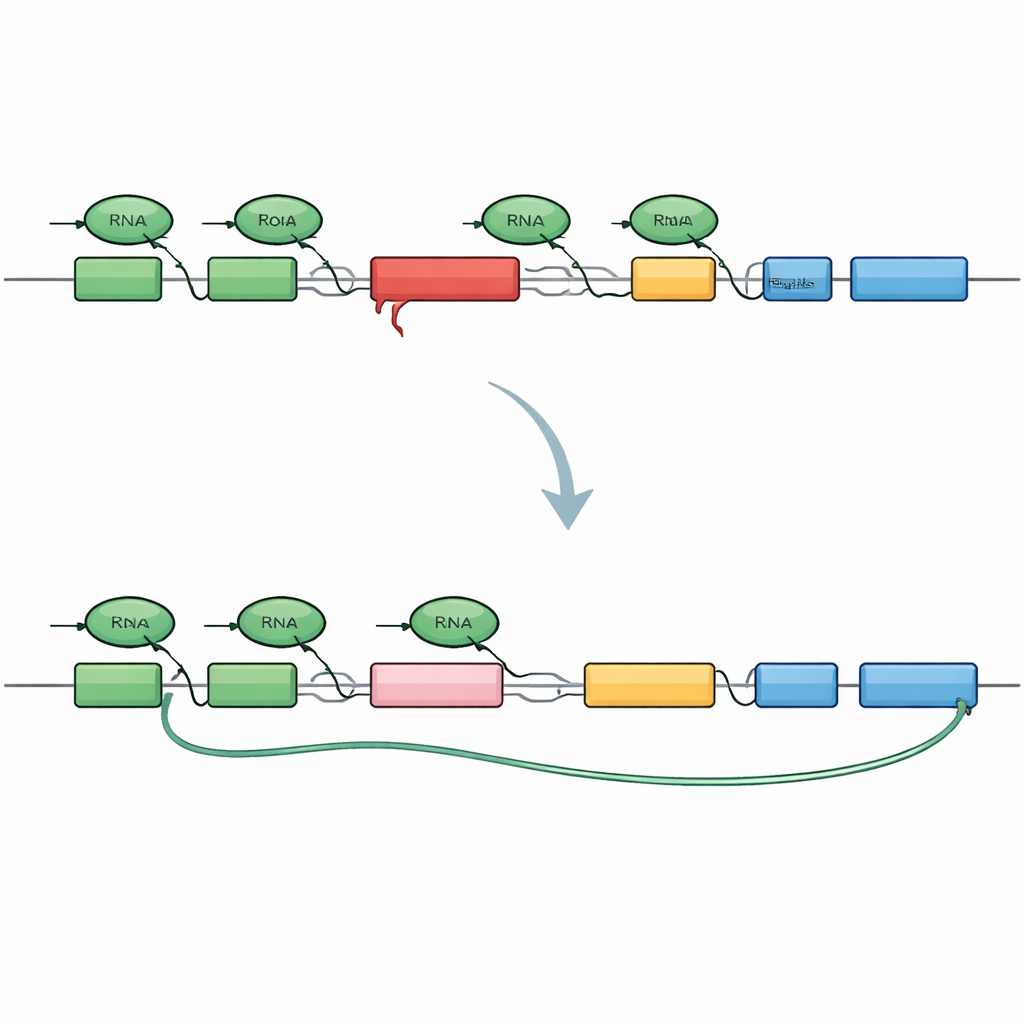
Aby rozłożyć problem na czynniki, badacze skonstruowali miniaturową wersję genu TAF1 zawierającą ten sam insert SVA występujący u pacjentów. Oznaczyli początek i koniec mini‑genu różnymi barwnikami fluorescencyjnymi, tak że prawidłowe przepisywanie zapalało oba końce, podczas gdy przedwczesne przerwanie świeciło tylko na początku. Gdy konstrukcja ta została wprowadzona do komórek ludzkich, wiernie odtworzyła wzorzec XDP: wiele transkryptów kończyło się przedwcześnie w intronie zawierającym SVA, a ukryty „kryptyczny” fragment sekwencji był włączany do splicingu, tworząc skrócone, ślepe końcówki mRNA i zredukowane fragmenty białek. Potwierdziło to, że sam insert SVA wystarcza, by przekształcić intron w silną przeszkodę dla aparatu transkrypcyjnego.
Zmniejszenie BRD4 pozwala odczytać komunikaty dalej
Posiadając ten sensor w żywych komórkach, zespół przesiał ponad 500 małych cząsteczek celujących w szlaki kontroli genów i chromatyny, szukając związków, które zwiększały sygnał fluorescencyjny z dalekiego końca mini‑genu. Wyraźna grupa trafień działała poprzez degradowanie rodziny białek BET, przy czym BRD4 wysunął się na pierwszy plan. Gdy BRD4 został zredukowany, aparat transkrypcyjny częściej czytał przez problematyczny intron, pomijał kryptyczne miejsca splicingu i produkował pełnej długości transkrypty TAF1. Co zaskakujące, samo blokowanie klasycznych miejsc wiążących chromatynę BRD4 nie wystarczało; odpowiedzialny okazał się inny ogonowy region BRD4, wcześniej powiązany z kontrolą kończenia transkrypcji. To ujawnia niekanoniczną rolę BRD4 w zarządzaniu miejscami i sposobem przycinania oraz finalizowania komunikatów.
Miniaturowe mózgi ujawniają wczesne defekty tkankowe i częściową poprawę
Aby sprawdzić, czy ten mechanizm ma znaczenie w bardziej realistycznym modelu, autorzy hodowali trójwymiarowe organoidy przypominające mózg z komórek macierzystych pacjentów. Te maleńkie „mini‑mózgi” naśladują wczesny rozwój ludzkiego mózgu. Organoidy niosące mutację XDP były mniejsze i wykazywały zdeformowane warstwy komórek macierzystych neuralnych i ich potomstwa oraz zwiększoną śmiertelność komórek, co wskazuje na wczesny stres rozwojowy. Przy użyciu sekwencjonowania długiego odczytu zespół wykrył tę samą rodzinę przedwcześnie zakończonych transkryptów TAF1, obserwowaną w prostszych modelach komórkowych, a nawet w tkance mózgowej pacjentów pośmiertnie. Krótkotrwałe leczenie organoidów XDP degraderem ukierunkowanym na BRD4 przesunęło wzorce splicingu z dala od kryptycznych egzonów intronowych i nieco zwiększyło wykorzystanie normalnego egzagonu dalszego, wskazując na częściowe przywrócenie prawidłowego przetwarzania komunikatów.
Co to oznacza dla przyszłych pomysłów terapeutycznych
Mówiąc prościej, XDP można postrzegać jako problem, w którym zakłócający insert DNA zamienia długi gen w pole minowe fałszywych sygnałów stopu, powodując, że komórka produkuje okaleczone instrukcje. To badanie pokazuje, że obniżenie aktywności BRD4 może pomóc maszynerii transkrypcyjnej zignorować te fałszywe przerwy i dojść do właściwego końca genu, przywracając bardziej kompletne instrukcje. Chociaż obecne leki degradowalne skierowane przeciw BRD4 są zbyt nieprecyzyjne i toksyczne do bezpośredniego stosowania u pacjentów, ujawniają, że faza „linii mety” przepisywania jest wrażliwym i regulowalnym etapem. Celowanie w precyzyjnych partnerów i działania BRD4 zaangażowane w przycinanie komunikatów mogłoby w przyszłości umożliwić korekcję molekularnych defektów XDP, a być może także innych chorób spowodowanych wadliwym przetwarzaniem RNA.
Cytowanie: Capponi, S., Ehret, S., Camgöz, Z. et al. Correction of the molecular phenotype of X-linked Dystonia-Parkinsonism reveals a non-canonical function of BRD4. Nat Commun 17, 4062 (2026). https://doi.org/10.1038/s41467-026-72319-6
Słowa kluczowe: Dystonia‑Parkinsonizm sprzężony z chromosomem X, przetwarzanie RNA, BRD4, gen TAF1, retrotranspozon SVA