Clear Sky Science · de
Korrektur des molekularen Phänotyps bei X‑chromosomaler Dystonie‑Parinsonismus zeigt eine nicht‑kanonische Funktion von BRD4
Warum diese Geschichte über eine Gehirnerkrankung wichtig ist
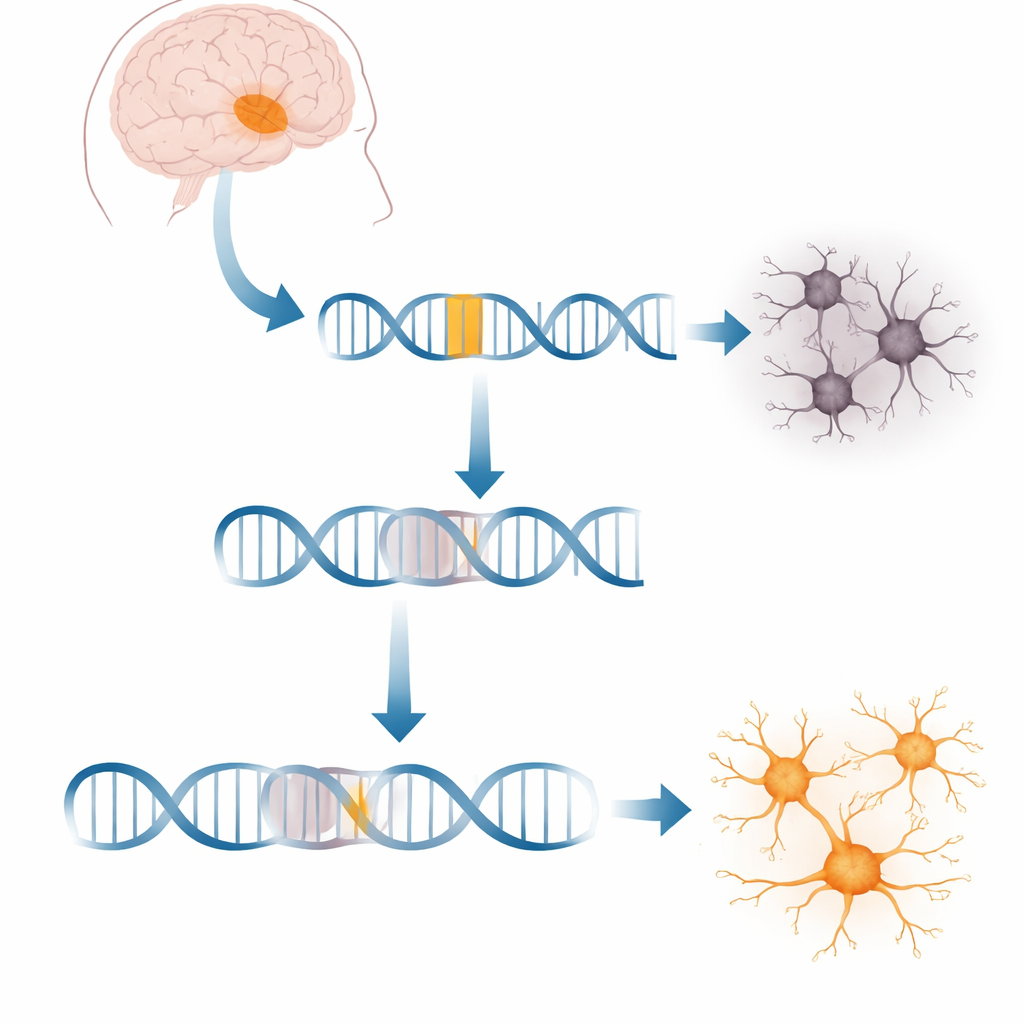
X‑chromosomaler Dystonie‑Parinsonismus (XDP) ist eine verheerende Bewegungsstörung, die vor allem Männer von den Philippinen trifft und verdrehende Bewegungen sowie Parkinson‑ähnliche Steifheit verursacht, die mit der Zeit schlechter werden. Obwohl Forschende die genetische Mutation kennen, die XDP auslöst, war bislang nicht genau klar, wie diese Veränderung die Nachrichten des Gens in Nervenzellen durcheinanderbringt — oder wie man das korrigieren könnte. Diese Studie legt eine verborgene Schwachstelle in der Art offen, wie Gehirnzellen genetische Nachrichten kopieren und beenden, und zeigt, dass das Herunterregeln eines bekannten regulatorischen Proteins, BRD4, die Nachrichten in Richtung Normalzustand zurückführen kann. Die Arbeit eröffnet eine neue Sicht auf Behandlung: Anstatt nur zu versuchen, fehlendes Protein zu ersetzen, könnte man möglicherweise die Verarbeitung der Nachricht selbst reparieren.
Ein seltsamer genetischer Einschub, der ein zentrales Hirngen entgleisen lässt
Jede Zelle kopiert ständig Gene in Botenmoleküle, die die Proteinproduktion steuern. Bei XDP ist ein Abschnitt beweglicher DNA, ein sogenanntes SVA‑Element, in ein großes Gen namens TAF1 gesprungen, das dabei hilft, das Ablesen von Tausenden anderer Gene zu starten. Dieser Einschub liegt tief in einem der langen nicht‑kodierenden Abschnitte (Introns) von TAF1. Die Zellen von Patientinnen und Patienten produzieren etwa 20 % weniger voll‑ständige TAF1‑Botschaften, was Netzwerke von Genen stört, die für gesunde Neurone notwendig sind. Unklar war bislang, wie genau diese fremde Sequenz den Kopiervorgang unterbricht und ob man Zellen überreden kann, diese Blockade zu umgehen.
Ein lebender Sensor für fehlerhafte Genbotschaften
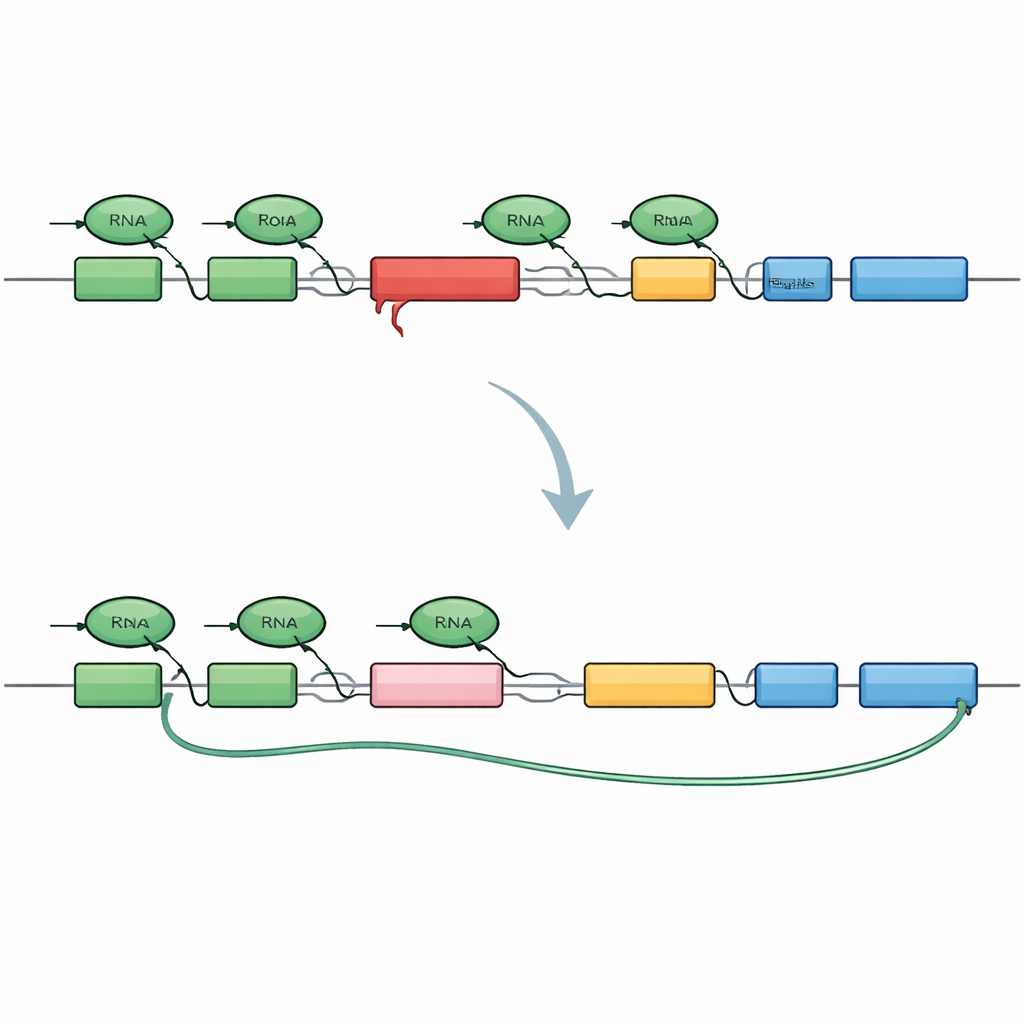
Um das Problem zu untersuchen, bauten die Forschenden eine verkleinerte Version des TAF1‑Gens nach, die denselben SVA‑Einschub wie bei Patientinnen und Patienten enthält. Sie markierten Anfang und Ende dieses Mini‑Gens mit unterschiedlichen fluoreszierenden Farben, sodass bei intaktem Ablesen beide Enden aufleuchten, während bei vorzeitigem Abbruch nur der Anfang sichtbar wäre. Als dieses Konstrukt in menschliche Zellen eingebracht wurde, reproduzierte es treu das XDP‑Muster: Viele Nachrichten endeten vorzeitig innerhalb des Introns, das das SVA trägt, und ein verborgenes „kryptisches“ Sequenzstück wurde eingebaut, wodurch verkürzte, Sackgassen‑Botschaften und abgeschnittene Proteinfragmente entstanden. Das bestätigte, dass das SVA allein ausreicht, um das Intron zu einer starken Blockade für die Kopiermaschinerie zu machen.
BRD4‑Herunterregeln erlaubt das Durchlesen der Botschaften
Mit diesem Live‑Zell‑Sensor durchsuchte das Team mehr als 500 kleine Moleküle, die Genkontroll‑ und Chromatinwege ansprechen, und suchte nach Verbindungen, die das fluoreszierende Signal am entfernten Ende des Mini‑Gens verstärkten. Eine auffällige Gruppe von Treffern wirkte, indem sie eine Familie von Proteinen, die BET‑Proteine, abbauten; BRD4 trat dabei als Schlüsselspieler hervor. Wurde BRD4 vermindert, las die Kopiermaschinerie häufiger über das problematische Intron hinweg, übersprang die kryptischen Spleißstellen und produzierte voll‑ständige TAF1‑Botschaften. Überraschenderweise reichte es nicht aus, nur die bekannten Chromatinbindungsstellen von BRD4 zu blockieren; stattdessen war eine andere Schwanzregion von BRD4 verantwortlich, die zuvor mit der Kontrolle des Transkriptionsendes in Verbindung gebracht worden war. Das zeigt eine nicht‑kanonische Rolle von BRD4 bei der Steuerung, wie und wo Botschaften geschnitten und beendet werden.
Mini‑Gehirne enthüllen frühe Gewebedefekte und partielle Rettung
Um zu prüfen, ob dieser Mechanismus in einem realistischeren Kontext relevant ist, züchteten die Autorinnen und Autoren dreidimensionale gehirnähnliche Organoide aus Stammzellen der Patientinnen und Patienten. Diese winzigen „Mini‑Gehirne“ ahmen frühe menschliche Gehirnentwicklung nach. Organoide mit der XDP‑Mutation waren kleiner und zeigten verzerrte Schichten aus neuralen Stammzellen und deren Nachkommen sowie erhöhte Zellsterblichkeit, was auf frühen Entwicklungsstress hindeutet. Mittels Long‑Read‑Sequenzierung detektierte das Team dieselbe Familie vorzeitig beendeter TAF1‑Botschaften, die bereits in einfacheren Zellmodellen und sogar in postmortem gewonnenem Patienten‑Gehirngewebe zu sehen war. Eine Kurzzeitbehandlung der XDP‑Organoide mit einem BRD4‑gerichteten Degrader verschob die Spleißmuster weg von kryptischen intronischen Exons und erhöhte moderat die Nutzung des normalen nachgeschalteten Exons, was auf eine teilweise Wiederherstellung einer gesunden Nachrichtenverarbeitung hindeutet.
Was das für künftige Behandlungsansätze bedeutet
Anschaulich lässt sich XDP als ein Problem verstehen, bei dem ein störender DNA‑Einschub ein langes Gen in ein Minenfeld falscher Stopp‑Signale verwandelt, wodurch die Zelle verstümmelte Anweisungen produziert. Diese Studie zeigt, dass eine Verringerung der BRD4‑Aktivität der Kopiermaschinerie helfen kann, diese falschen Stopps zu ignorieren und bis zum richtigen Ende des Gens weiterzulesen, wodurch vollständigere Anweisungen wiederhergestellt werden. Obwohl die derzeitigen BRD4‑abbauenden Wirkstoffe viel zu unspezifisch und toxisch für den direkten Einsatz beim Menschen sind, machen sie deutlich, dass die „Ziellinie“ des Genablesens eine verwundbare und verstellbare Stufe ist. Das gezielte Ansteuern der spezifischen Partner und Funktionen von BRD4, die an der Botschafts‑Beendigung beteiligt sind, könnte künftig einen Weg bieten, die molekularen Defekte von XDP und möglicherweise anderer durch fehlerhafte RNA‑Verarbeitung bedingter Erkrankungen zu korrigieren.
Zitation: Capponi, S., Ehret, S., Camgöz, Z. et al. Correction of the molecular phenotype of X-linked Dystonia-Parkinsonism reveals a non-canonical function of BRD4. Nat Commun 17, 4062 (2026). https://doi.org/10.1038/s41467-026-72319-6
Schlüsselwörter: X‑chromosomaler Dystonie‑Parinsonismus, RNA‑Verarbeitung, BRD4, TAF1‑Gen, SVA‑Retrotransposon