Clear Sky Science · en
Correction of the molecular phenotype of X-linked Dystonia-Parkinsonism reveals a non-canonical function of BRD4
Why this brain disorder story matters
X‑linked Dystonia‑Parkinsonism (XDP) is a devastating movement disorder that strikes mainly men from the Philippines, causing twisting movements and Parkinson‑like stiffness that worsen over time. Although scientists know the genetic mutation that triggers XDP, they have not understood exactly how this change scrambles the gene’s messages inside nerve cells — or how to correct it. This study uncovers a hidden weak point in the way brain cells copy and finish genetic messages and shows that dialing down a well‑known regulatory protein, BRD4, can restore more normal messages. The work opens up a fresh way of thinking about treatment: instead of only trying to replace missing protein, we might be able to repair how the message itself is processed.
A strange genetic insert that derails a key brain gene
Every cell constantly copies genes into messenger molecules that guide protein production. In XDP, a stretch of mobile DNA called an SVA element has jumped into a large gene named TAF1, which helps launch the reading of thousands of other genes. This insert sits deep inside one of TAF1’s long non‑coding stretches. Patients’ cells make about 20% less full‑length TAF1 message, disturbing networks of genes needed for healthy neurons. What was unclear is exactly how this foreign sequence interrupts the copying process and whether cells can be coaxed to bypass the blockage.
Building a living sensor for faulty gene messages
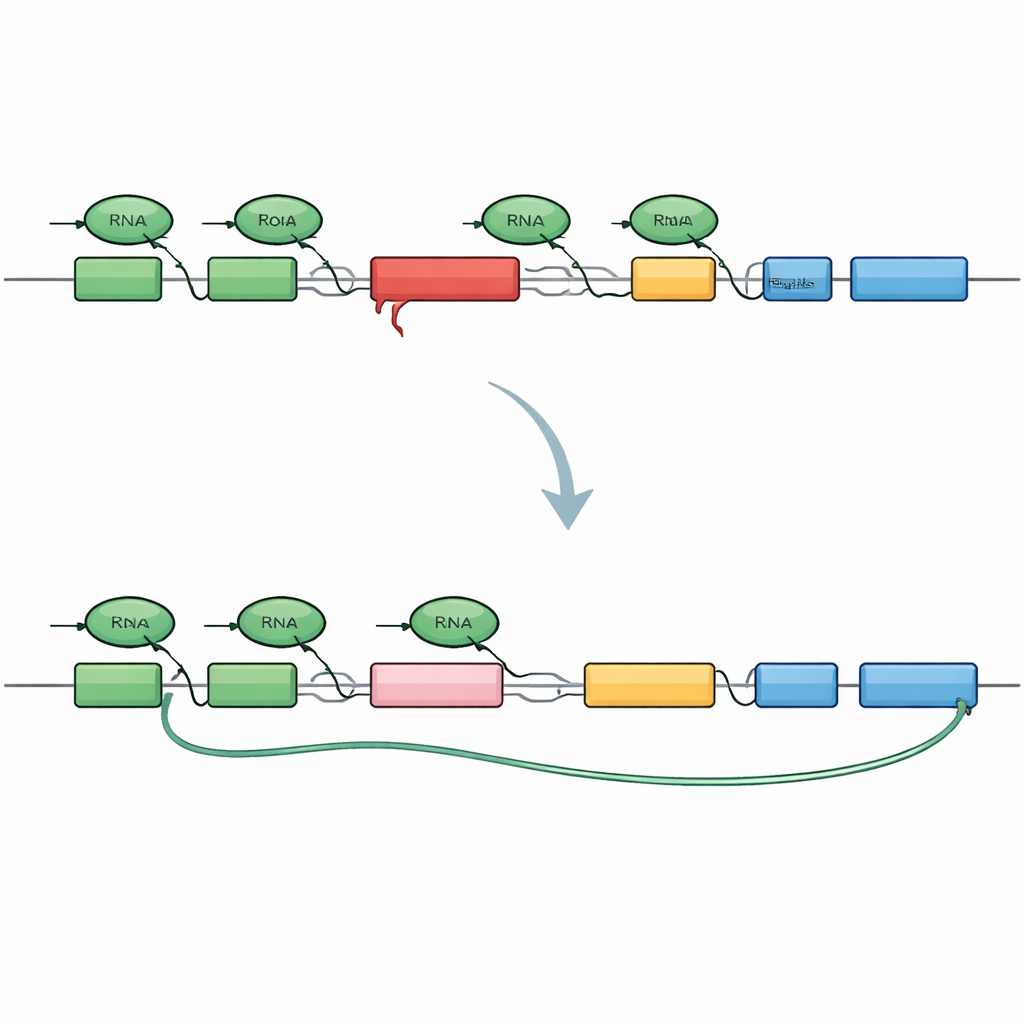
To dissect the problem, the researchers engineered a miniature version of the TAF1 gene that includes the same SVA insert found in patients. They tagged the beginning and end of this mini‑gene with different fluorescent colors so that healthy copying would light up both ends, whereas early cut‑off would light only the start. When this construct was placed into human cells, it faithfully reproduced the XDP pattern: many messages stopped prematurely within the intron that carries the SVA, and a hidden “cryptic” piece of sequence was spliced in, creating shortened, dead‑end messages and truncated protein fragments. This confirmed that the SVA alone is enough to turn the intron into a strong roadblock for the copying machinery.
Turning down BRD4 lets messages read through
Armed with this live‑cell sensor, the team screened more than 500 small molecules that target gene‑control and chromatin pathways, looking for compounds that boosted the fluorescent signal from the mini‑gene’s far end. A striking cluster of hits all worked by degrading a family of proteins called BET proteins, with BRD4 emerging as the main player. When BRD4 was depleted, the copying machinery was more likely to read past the problematic intron, skip the cryptic splice sites and produce full‑length TAF1 messages. Surprisingly, simply blocking BRD4’s usual chromatin‑binding pockets was not enough; instead, a different tail region of BRD4, previously linked to controlling the end of transcription, was responsible. This reveals a non‑canonical role for BRD4 in managing how and where messages are cut and finished.
Miniature brains reveal early tissue defects and partial rescue
To test whether this mechanism matters in a more realistic setting, the authors grew three‑dimensional brain‑like organoids from patients’ stem cells. These tiny “mini‑brains” mimic early human brain development. Organoids carrying the XDP mutation were smaller and showed distorted layers of neural stem cells and their progeny, along with increased cell death, pointing to early developmental stress. Using long‑read sequencing, the team detected the same family of prematurely terminated TAF1 messages seen in simpler cell models and even in postmortem patient brain tissue. Short‑term treatment of XDP organoids with a BRD4‑targeting degrader shifted splicing patterns away from cryptic intronic exons and modestly increased use of the normal downstream exon, indicating a partial restoration of healthy message processing.
What this means for future treatment ideas
In everyday terms, XDP can be viewed as a problem in which a disruptive DNA insert turns a long gene into a minefield of false stop signals, causing the cell to churn out stunted instructions. This study shows that lowering BRD4 activity can help the cellular copying machinery ignore those false stops and continue to the proper end of the gene, restoring more complete instructions. Although the current BRD4‑degrading drugs are far too blunt and toxic for direct use in patients, they reveal that the “finish line” phase of gene copying is a vulnerable and adjustable step. Targeting the precise partners and actions of BRD4 involved in message trimming could, in the future, offer a way to correct the molecular defects of XDP and perhaps other diseases driven by faulty RNA processing.
Citation: Capponi, S., Ehret, S., Camgöz, Z. et al. Correction of the molecular phenotype of X-linked Dystonia-Parkinsonism reveals a non-canonical function of BRD4. Nat Commun 17, 4062 (2026). https://doi.org/10.1038/s41467-026-72319-6
Keywords: X-linked Dystonia-Parkinsonism, RNA processing, BRD4, TAF1 gene, SVA retrotransposon