Clear Sky Science · es
Corrección del fenotipo molecular de la distonía‑Parkinson ligada al cromosoma X revela una función no canónica de BRD4
Por qué importa esta historia sobre un trastorno cerebral
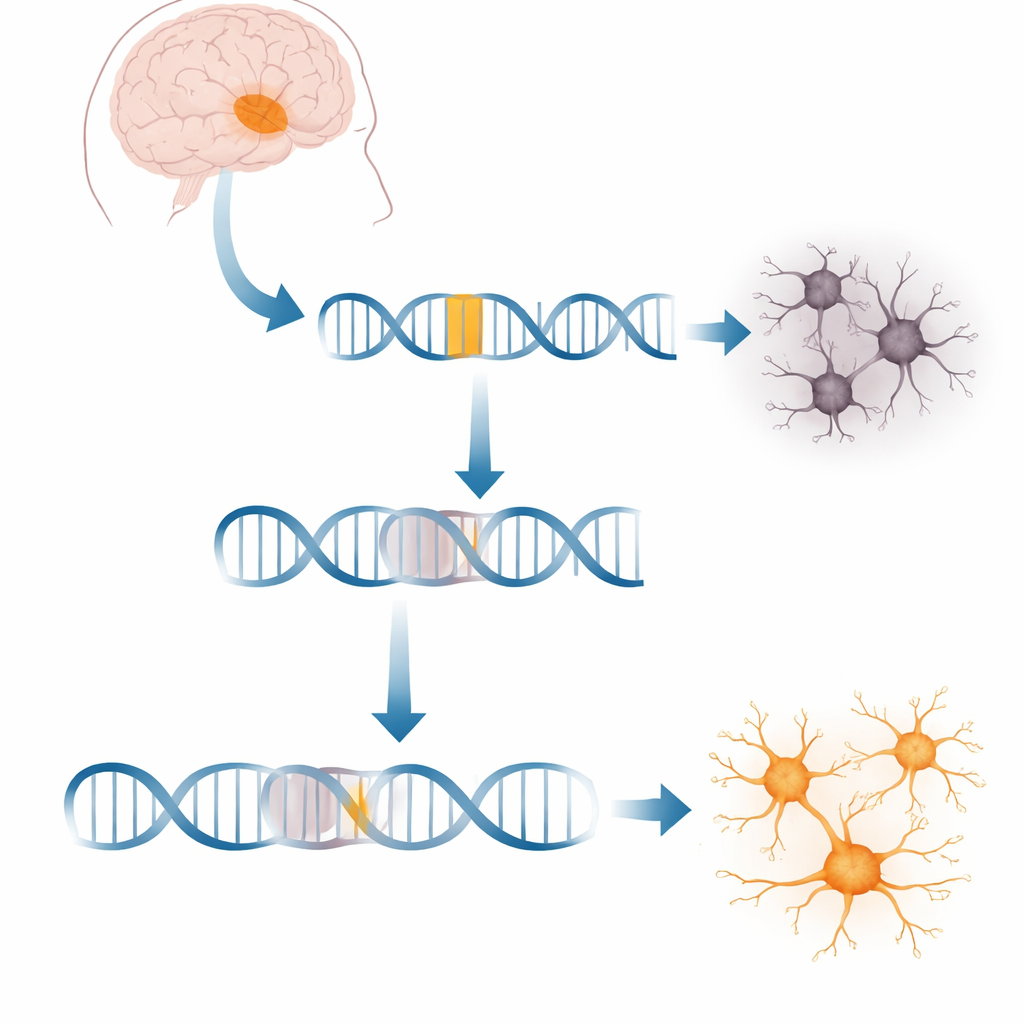
La distonía‑Parkinson ligada al cromosoma X (XDP) es un devastador trastorno del movimiento que afecta principalmente a hombres de Filipinas, provocando movimientos torsionales y rigidez similar al Parkinson que empeoran con el tiempo. Aunque los científicos conocen la mutación genética que desencadena la XDP, no habían entendido exactamente cómo ese cambio desordena los mensajes del gen dentro de las neuronas ni cómo corregirlo. Este estudio descubre un punto débil oculto en la manera en que las células cerebrales copian y finalizan los mensajes genéticos y muestra que reducir la actividad de una proteína reguladora bien conocida, BRD4, puede restaurar mensajes más normales. El trabajo abre una nueva forma de pensar en el tratamiento: en lugar de limitarse a intentar reemplazar la proteína faltante, podríamos reparar cómo se procesa el propio mensaje.
Una extraña inserción genética que descarrila un gen cerebral clave
Cada célula copia constantemente los genes en moléculas mensajeras que guían la producción de proteínas. En la XDP, un tramo de ADN móvil llamado elemento SVA se ha insertado en un gen grande denominado TAF1, que ayuda a iniciar la lectura de miles de otros genes. Esta inserción se sitúa en el interior de uno de los largos tramos no codificantes de TAF1. Las células de los pacientes producen alrededor de un 20 % menos del mensaje completo de TAF1, alterando redes de genes necesarias para neuronas saludables. Lo que no estaba claro es exactamente cómo esta secuencia foránea interrumpe el proceso de copia y si se puede inducir a las células a sortear ese bloqueo.
Construir un sensor vivo para mensajes genéticos defectuosos
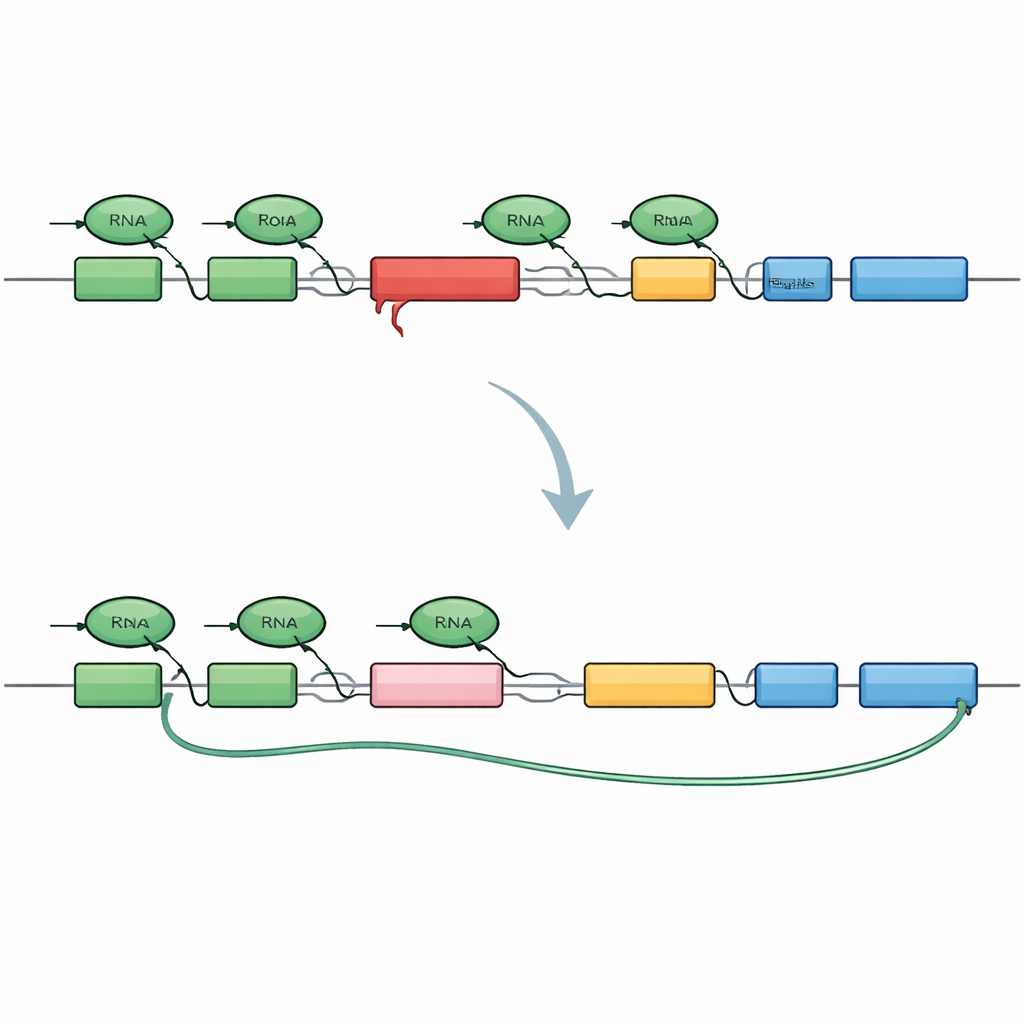
Para analizar el problema, los investigadores diseñaron una versión miniaturizada del gen TAF1 que incluye la misma inserción SVA encontrada en los pacientes. Marcaban el principio y el final de este mini‑gen con diferentes colores fluorescentes de modo que una copia sana iluminara ambos extremos, mientras que un corte prematuro solo iluminaría el inicio. Cuando este constructo se introdujo en células humanas, reprodujo fielmente el patrón de la XDP: muchos mensajes se detenían prematuramente dentro del intrón que contiene el SVA, y una pieza oculta «críptica» de la secuencia se empalmaba, creando mensajes acortados sin salida y fragmentos proteicos truncados. Esto confirmó que el SVA por sí solo basta para convertir el intrón en un fuerte obstáculo para la maquinaria de copia.
Reducir BRD4 permite que los mensajes se lean hasta el final
Armado con este sensor en células vivas, el equipo cribó más de 500 pequeñas moléculas que actúan sobre vías de control génico y cromatina, buscando compuestos que aumentaran la señal fluorescente del extremo lejano del mini‑gen. Un grupo notable de compuestos actuó degradando una familia de proteínas llamadas BET, con BRD4 emergiendo como el actor principal. Cuando BRD4 se agotó, la maquinaria de transcripción tuvo más probabilidades de leer más allá del intrón problemático, evitar los sitios de empalme crípticos y producir mensajes completos de TAF1. Sorprendentemente, bloquear simplemente las habituales bolsas de unión a cromatina de BRD4 no fue suficiente; en su lugar, una región de cola distinta de BRD4, previamente vinculada al control del final de la transcripción, fue la responsable. Esto revela un papel no canónico de BRD4 en la gestión de cómo y dónde se cortan y finalizan los mensajes.
Cerebros en miniatura revelan defectos tisulares tempranos y rescate parcial
Para comprobar si este mecanismo tiene repercusión en un entorno más realista, los autores cultivaron organoides cerebrales tridimensionales a partir de células madre de pacientes. Estos pequeños «mini‑cerebros» imitan el desarrollo cerebral humano temprano. Los organoides portadores de la mutación XDP eran más pequeños y mostraban capas distorsionadas de células madre neurales y sus progenie, junto con un aumento de la muerte celular, señalando estrés en las primeras fases del desarrollo. Mediante secuenciación de lectura larga, el equipo detectó la misma familia de mensajes de TAF1 terminados prematuramente que se observó en modelos celulares más simples e incluso en tejido cerebral postmortem de pacientes. El tratamiento a corto plazo de organoides XDP con un degradador dirigido a BRD4 cambió los patrones de empalme alejándolos de exones intrónicos crípticos e incrementó modestamente el uso del exón normal aguas abajo, indicando una restauración parcial del procesamiento saludable de los mensajes.
Qué significa esto para futuras ideas terapéuticas
En términos cotidianos, la XDP puede verse como un problema en el que una inserción de ADN disruptiva convierte un gen largo en un campo minado de señales de parada falsas, haciendo que la célula produzca instrucciones truncadas. Este estudio muestra que reducir la actividad de BRD4 puede ayudar a la maquinaria de copia celular a ignorar esas paradas falsas y continuar hasta el final correcto del gen, restaurando instrucciones más completas. Aunque los fármacos degradadores de BRD4 actuales son demasiado toscos y tóxicos para su uso directo en pacientes, revelan que la fase de «meta» de la transcripción es un paso vulnerable y ajustable. Dirigir a los socios y las acciones precisas de BRD4 implicadas en el recorte de mensajes podría, en el futuro, ofrecer una forma de corregir los defectos moleculares de la XDP y quizá de otras enfermedades impulsadas por un procesamiento erróneo del ARN.
Cita: Capponi, S., Ehret, S., Camgöz, Z. et al. Correction of the molecular phenotype of X-linked Dystonia-Parkinsonism reveals a non-canonical function of BRD4. Nat Commun 17, 4062 (2026). https://doi.org/10.1038/s41467-026-72319-6
Palabras clave: Distonía‑Parkinson ligada al cromosoma X, Procesamiento del ARN, BRD4, gen TAF1, retrotransposón SVA