Clear Sky Science · fr
Correction du phénotype moléculaire de la dystonie-parkinsonisme liée à l’X révèle une fonction non canonique de BRD4
Pourquoi cette histoire de maladie cérébrale compte
La dystonie‑parkinsonisme liée à l’X (XDP) est un trouble du mouvement dévastateur qui touche principalement les hommes originaires des Philippines, provoquant des mouvements de torsion et une raideur de type Parkinson qui s’aggravent avec le temps. Bien que les scientifiques connaissent la mutation génétique qui déclenche la XDP, ils n’avaient pas encore compris précisément comment ce changement perturbe les messages du gène au sein des cellules nerveuses — ni comment le corriger. Cette étude met au jour une faiblesse cachée dans la manière dont les cellules cérébrales copient et terminent les messages génétiques et montre que réduire l’activité d’une protéine régulatrice bien connue, BRD4, peut restaurer des messages plus normaux. Le travail ouvre une nouvelle perspective thérapeutique : plutôt que de se contenter de remplacer une protéine manquante, on pourrait réparer la façon dont le message lui‑même est traité.
Une insertion génétique étrange qui fait dérailler un gène cérébral clé
Chaque cellule copie en permanence les gènes en molécules messagères qui dirigent la production des protéines. Dans la XDP, un segment d’ADN mobile appelé élément SVA s’est inséré dans un grand gène nommé TAF1, qui aide à lancer la lecture de milliers d’autres gènes. Cet insert se trouve profondément à l’intérieur d’un des longs segments non codants de TAF1. Les cellules des patients produisent environ 20 % de message TAF1 complet en moins, perturbant des réseaux de gènes nécessaires au bon fonctionnement des neurones. Ce qui restait obscur, c’est comment cette séquence étrangère interrompait précisément le processus de copie et si les cellules pouvaient être amenées à contourner ce blocage.
Construire un capteur vivant pour les messages génétiques défectueux
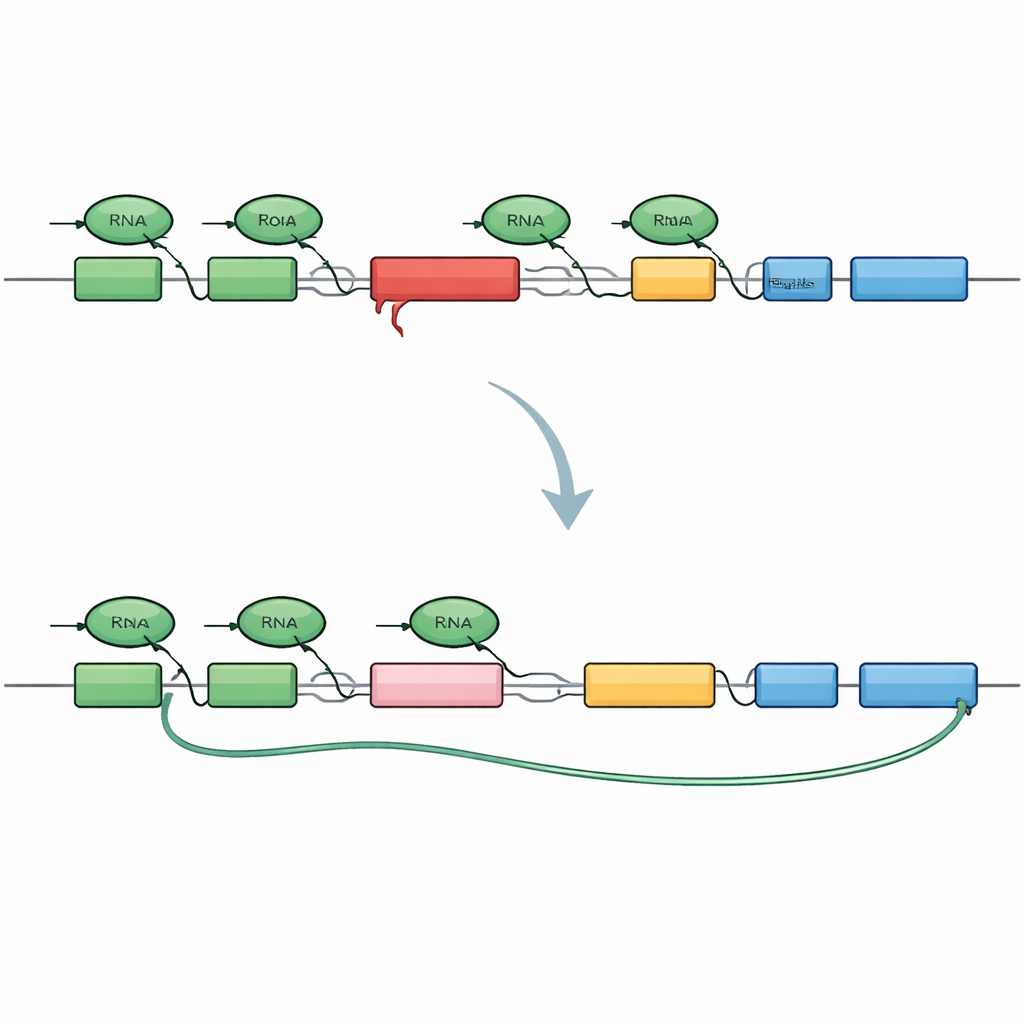
Pour analyser le problème, les chercheurs ont conçu une version miniature du gène TAF1 qui inclut le même insert SVA trouvé chez les patients. Ils ont marqué le début et la fin de ce mini‑gène par des couleurs fluorescentes différentes de sorte qu’une copie saine illuminerait les deux extrémités, tandis qu’une coupure précoce n’allumerait que le début. Lorsque ce construct a été placé dans des cellules humaines, il a fidèlement reproduit le profil de la XDP : de nombreux messages s’arrêtaient prématurément dans l’intron qui porte le SVA, et un fragment de séquence « cryptique » était épissé, créant des messages raccourcis et sans suite ainsi que des fragments protéiques tronqués. Cela a confirmé que le seul SVA suffit à transformer l’intron en un obstacle puissant pour la machinerie de copie.
Abaisser BRD4 permet la lecture au‑delà du blocage
Dotés de ce capteur en cellules vivantes, l’équipe a criblé plus de 500 petites molécules ciblant des voies de contrôle génique et de chromatine, à la recherche de composés augmentant le signal fluorescent provenant de l’extrémité distale du mini‑gène. Un groupe marquant de composés actifs agissait en dégradant une famille de protéines appelées protéines BET, BRD4 se distinguant comme l’acteur principal. Lorsque BRD4 était diminué, la machinerie de transcription lisait plus volontiers au‑delà de l’intron problématique, sautait les sites d’épissage cryptiques et produisait des messages TAF1 de longueur complète. De façon surprenante, bloquer simplement les domaines classiques de liaison à la chromatine de BRD4 ne suffisait pas ; c’est plutôt une région terminale différente de BRD4, liée auparavant au contrôle de la terminaison de la transcription, qui était responsable. Cela révèle un rôle non canonique de BRD4 dans la gestion du lieu et de la manière dont les messages sont coupés et terminés.
Mini‑cerveaux révèlent des défauts tissulaires précoces et une restauration partielle
Pour vérifier si ce mécanisme est pertinent dans un contexte plus réaliste, les auteurs ont cultivé des organoïdes cérébraux tridimensionnels à partir de cellules souches de patients. Ces « mini‑cerveaux » reproduisent le développement cérébral humain précoce. Les organoïdes porteurs de la mutation XDP étaient plus petits et présentaient des couches de cellules souches neurales et de leurs descendants déformées, ainsi qu’une augmentation de la mort cellulaire, suggérant un stress développemental précoce. Grâce au séquençage longue‑lecture, l’équipe a détecté la même famille de messages TAF1 prématurément terminés observée dans des modèles cellulaires plus simples et même dans des tissus cérébraux post‑mortem de patients. Un traitement à court terme des organoïdes XDP avec un dégradeur ciblant BRD4 a fait évoluer les schémas d’épissage loin des exons introniques cryptiques et a modestement augmenté l’utilisation de l’exon normal en aval, indiquant une restauration partielle du traitement des messages.
Ce que cela signifie pour les idées de traitement futures
En termes simples, la XDP peut être vue comme un problème où une insertion d’ADN perturbatrice transforme un long gène en un champ de mines de faux signaux d’arrêt, poussant la cellule à produire des instructions tronquées. Cette étude montre que réduire l’activité de BRD4 peut aider la machinerie à ignorer ces faux arrêts et à poursuivre jusqu’à la bonne fin du gène, restituant des instructions plus complètes. Bien que les médicaments actuels qui dégradent BRD4 soient encore trop grossiers et toxiques pour un usage direct chez les patients, ils démontrent que la phase de « ligne d’arrivée » de la transcription est une étape vulnérable et modulable. Cibler les partenaires précis et les actions de BRD4 impliqués dans le façonnage des messages pourrait, à l’avenir, offrir une manière de corriger les défauts moléculaires de la XDP et peut‑être d’autres maladies dues à un mauvais traitement de l’ARN.
Citation: Capponi, S., Ehret, S., Camgöz, Z. et al. Correction of the molecular phenotype of X-linked Dystonia-Parkinsonism reveals a non-canonical function of BRD4. Nat Commun 17, 4062 (2026). https://doi.org/10.1038/s41467-026-72319-6
Mots-clés: Dystonie‑Parkinsonisme liée à l’X, Traitement de l’ARN, BRD4, gène TAF1, rétrotransposon SVA