Clear Sky Science · pl
DUSP26 chroni przed ostrą niewydolnością nerek przez defosforylację p53 w serynie 312
Dlaczego stres nerek dotyczy nas wszystkich
Ostra niewydolność nerek to nagły spadek funkcji tego narządu, który może nastąpić podczas poważnej operacji, ciężkiej infekcji, odwodnienia lub leczenia onkologicznego. Jest częsta w szpitalach i zwiększa ryzyko konieczności dializ, dłuższego pobytu na OIOM-ie oraz późniejszego rozwoju przewlekłej choroby nerek. Mimo to lekarze wciąż nie dysponują ukierunkowanym lekiem, który bezpośrednio chroniłby komórki pracujące w nerkach. Badanie to odkrywa wbudowany przełącznik ochronny w tych komórkach i pokazuje, jak jego włączenie lub wyłączenie może radykalnie zmienić, czy nerki ulegną uszkodzeniu, czy zostaną oszczędzone.
Ukryty obrońca w komórkach nerek
Autorzy skupili się na drobnych kanalikach w nerkach, które filtrują i dopracowują skład krwi. Wykorzystując kilka dużych ludzkich zbiorów danych oraz nowoczesne narzędzia uczenia maszynowego, wytypowali gen o nazwie DUSP26, który konsekwentnie występował na niższym poziomie u osób z ostrą niewydolnością nerek w porównaniu z osobami o zdrowych nerkach. W badaniu próbek biopsji ludzkich stwierdzili, że komórki kanalików bliższych — wykonujące większość ciężkiej pracy nerki — miały zdecydowanie obniżone ilości białka DUSP26 podczas urazu, a ten spadek ściśle korelował z poważniejszym uszkodzeniem tkanek i gorszą funkcją nerek. To sugerowało, że DUSP26 normalnie pomaga tym komórkom znosić stres.
Jak tarcza nerek zostaje wyłączona
Aby zrozumieć, dlaczego poziomy DUSP26 spadają, zespół sięgnął po modele mysie dwóch powszechnych form stresu nerkowego: lek chemioterapeutyczny toksyczny dla nerek oraz okres zablokowanego przepływu krwi, po którym następuje reperfuzja. W obu przypadkach poziomy DUSP26 w nerkach spadały równolegle ze wzrostem śmierci komórek. Ustalili, że ten spadek wynika z chemicznych znaczników, zwanych metylacją DNA, umieszczanych w regionie kontrolnym genu DUSP26. Znaczniki te działają jak taśma na przełączniku światła, uniemożliwiając włączenie genu. Eksperymenty na hodowlach komórek nerkowych wykazały, że uraz zwiększa metylację i wycisza DUSP26, a enzymy odpowiadające za dodawanie tych znaczników są rekrutowane do promotora genu. U myszy z urazem niedokrwienno‑reperfuzyjnym głębokie sekwencjonowanie potwierdziło, że wiele miejsc w tym samym regionie promotora staje się silnie zmetylowanych.
Wyłączanie i włączanie ochrony u zwierząt
Naukowcy zapytali następnie, co się stanie, jeśli tę molekularną tarczę usuniemy lub wzmocnimy. W hodowlach, blokowanie DUSP26 za pomocą narzędzi genetycznych lub małocząsteczkowego leku prowadziło do większej aktywacji kaspazy‑3, kluczowego egzekutora zaprogramowanej śmierci komórkowej, po stresie toksycznym lub niedotlenieniu. Zwiększenie poziomu DUSP26 miało efekt odwrotny, zmniejszając śmierć komórek. U myszy, wstępne podanie inhibitora DUSP26 przed uszkodzeniem nerek powodowało wyższe stężenia produktów przemiany azotowej we krwi, więcej mikroskopowych uszkodzeń i więcej obumierających komórek kanalików. Natomiast myszy zaprojektowane tak, by nadmiernie wytwarzać DUSP26 specyficznie w kanalikach bliższych, wykazały wyraźną ochronę przed zarówno uszkodzeniem wywołanym lekiem, jak i niedokrwiennym, z lepszą funkcją nerek i mniejszym uszkodzeniem tkanek. Te eksperymenty zwiększenia i utraty funkcji razem ujawniają DUSP26 jako potężnego strażnika integralności kanalików.
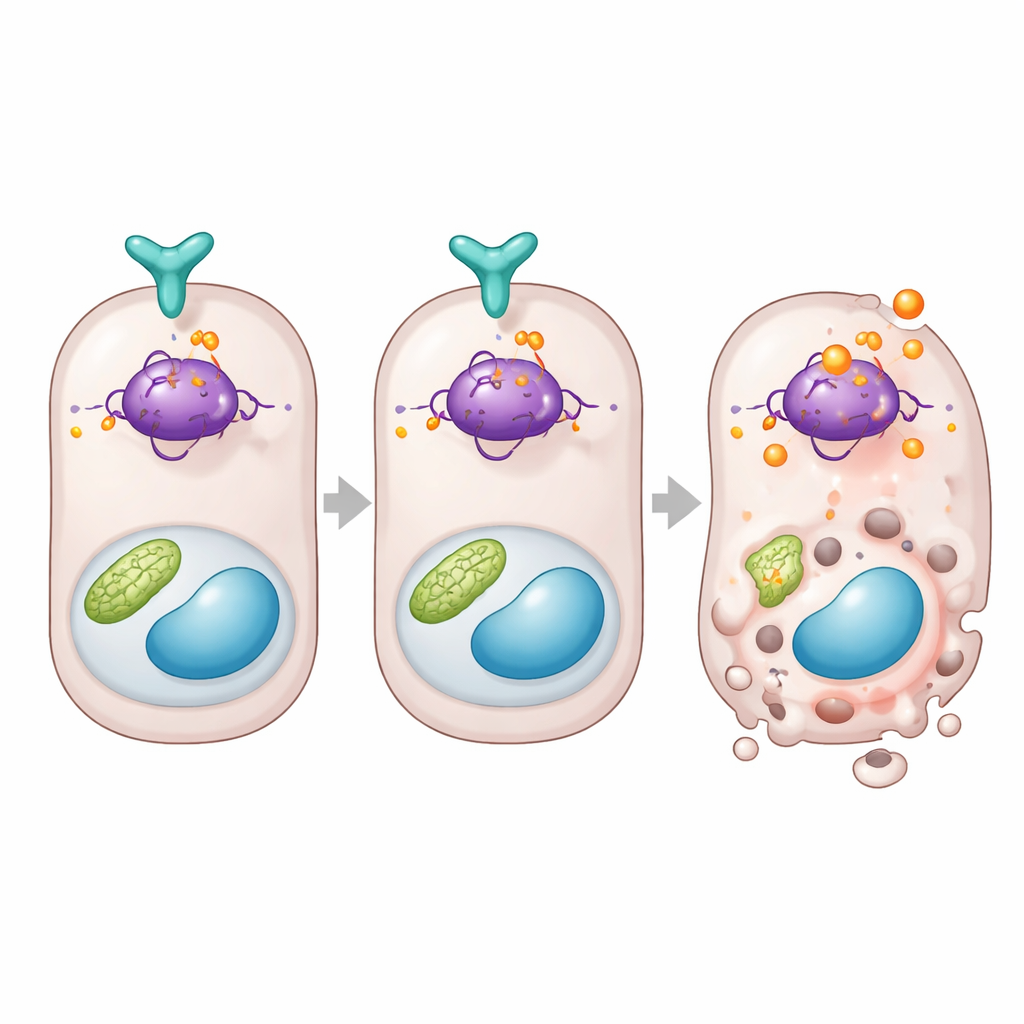
Bezpośredni hamulec dla białka decydującego o śmierci komórki
Bardziej wnikliwie, badanie łączy DUSP26 z p53, znanym białkiem decydującym, czy zestresowana komórka naprawi się, czy ulegnie samozniszczeniu. Zespół odkrył, że gdy DUSP26 było niskie w danych ludzkich lub nerkach myszy, geny kontrolowane przez p53 związane ze śmiercią, takie jak Bax i Puma, były zwiększone. W komórkach DUSP26 fizycznie wiązało się z p53 i selektywnie usuwało grupę fosforanową z jednego konkretnego miejsca na białku, znanego jako seryna 312 u myszy. Gdy DUSP26 było nieobecne, to miejsce stało się silnie zmodyfikowane, aktywność p53 wzrosła, a geny promujące śmierć zostały włączone. Aby udowodnić znaczenie tego miejsca, autorzy przebudowali komórki nerkowe i myszy z wersją p53, która nie mogła już być modyfikowana w tym punkcie. Te komórki i zwierzęta były znacznie bardziej odporne na uszkodzenie nerek, z mniejszą liczbą obumierających komórek kanalików i mniejszą dysfunkcją narządu, mimo że pozostałe elementy układu p53 pozostały nienaruszone.
Od wniknięcia molekularnego do przyszłych terapii
Podsumowując, praca opisuje jasny łańcuch zdarzeń: stres nerek zwiększa metylację DNA w genie DUSP26, obniżając poziom białka DUSP26 w kanalikach bliższych. Przy mniejszej ilości DUSP26 p53 pozostaje ufosforylowane w krytycznym miejscu, staje się bardziej aktywne i napędza ekspresję genów skłaniających komórki do śmierci. Przywrócenie DUSP26 lub naśladowanie jego działania na tym pojedynczym miejscu tłumi ten program śmierci i chroni tkankę nerkową. Dla pacjentów sugeruje to, że starannie zaprojektowane leki lub podejścia ukierunkowane genetycznie zwiększające aktywność DUSP26 — lub specyficznie zmniejszające fosforylację p53 w tym miejscu — mogłyby pewnego dnia pomóc chronić nerki podczas operacji, chemioterapii czy ciężkiej choroby, a być może także chronić inne narządy przed podobnymi formami ostrego uszkodzenia.
Cytowanie: Fu, Y., Xiang, Y., Han, Y. et al. DUSP26 protects against acute kidney injury by dephosphorylating p53 at serine 312. Nat Commun 17, 3208 (2026). https://doi.org/10.1038/s41467-026-69688-3
Słowa kluczowe: ostra niewydolność nerek, komórki kanalików nerkowych, szlak p53, regulacja epigenetyczna, śmierć komórkowa