Clear Sky Science · de
DUSP26 schützt vor akutem Nierenversagen durch Dephosphorylierung von p53 an Serin 312
Warum Nierenstress für alle wichtig ist
Akutes Nierenversagen ist ein plötzlicher Funktionsverlust der Niere, der bei größeren Operationen, schweren Infektionen, Dehydratation oder Krebstherapien auftreten kann. Es ist in Krankenhäusern häufig und erhöht das Risiko, eine Dialyse zu benötigen, länger auf der Intensivstation bleiben zu müssen und später eine chronische Nierenerkrankung zu entwickeln. Trotzdem gibt es bisher kein gezieltes Medikament, das direkt die arbeitenden Zellen der Niere schützt. Diese Studie zeigt einen eingebauten Schutzschalter in diesen Zellen und demonstriert, wie das An- oder Abschalten dieses Schalters entscheidend beeinflusst, ob Nieren geschädigt werden oder verschont bleiben.
Ein verborgener Verteidiger in Nierenzellen
Die Autoren konzentrierten sich auf die winzigen Tubuli in der Niere, die unser Blut filtern und feinabstimmen. Durch Auswertung mehrerer großer humaner Datensätze mit modernen Methoden des maschinellen Lernens identifizierten sie ein Gen namens DUSP26, das bei Menschen mit akutem Nierenversagen durchgängig niedriger exprimiert war als bei Gesunden. In menschlichen Biopsien fanden sie, dass proximalen Tubuluszellen – den Zellen, die den Großteil der Arbeit der Niere leisten – während der Verletzung deutlich geringere Mengen des DUSP26-Proteins hatten; dieser Verlust korrelierte eng mit stärkerer Gewebeschädigung und schlechterer Nierenfunktion. Das deutet darauf hin, dass DUSP26 diese Zellen normalerweise hilft, Stress zu überstehen.
Wie der Nierenschutz abgeschaltet wird
Um zu verstehen, warum die DUSP26-Spiegel fallen, nutzte das Team Mausmodelle von zwei gängigen Formen von Nierenstress: ein chemotherapeutisches Mittel mit Nierentoxizität und eine Phase unterbrochener Durchblutung gefolgt von Reperfusion. In beiden Situationen fielen die DUSP26-Spiegel in der Niere parallel zum Anstieg des Zelltods. Sie führten diesen Verlust auf chemische Markierungen zurück, sogenannte DNA-Methylierungen, die über den Kontrollbereich des DUSP26-Gens gelegt wurden. Diese Markierungen wirken wie Klebestreifen über einem Lichtschalter und verhindern, dass das Gen eingeschaltet wird. Experimente in kultivierten Nierenzellen zeigten, dass Verletzung die Methylierung erhöhte und DUSP26 ausschaltete und dass Enzyme, die diese Markierungen anbringen, an den Promotor des Gens gebunden wurden. In Mäusen mit Ischämie-Reperfusionsschaden bestätigte Tiefensequenzierung, dass viele Stellen innerhalb desselben Promotorbereichs stark methyliert wurden.
Schutz in Tieren aus- und einschalten
Die Forschenden fragten dann, was passiert, wenn dieser molekulare Schutz entfernt oder verstärkt wird. In Zellkulturen führte die Blockade von DUSP26 mittels genetischer Werkzeuge oder eines niedermolekularen Wirkstoffs nach toxischem oder sauerstoffarmem Stress zu stärkerer Aktivierung von Caspase‑3, einem zentralen Ausführer des programmierten Zelltods. Die Erhöhung von DUSP26 hatte den gegenteiligen Effekt und verringerte den Zelltod. In Mäusen führte die Vorbehandlung mit dem DUSP26-Inhibitor vor Nierenschädigung zu höheren Blutmesswerten für Abfallstoffe, mehr mikroskopischen Schäden und mehr sterbenden Tubuluszellen. Im Gegensatz dazu zeigten Mäuse, die so verändert waren, dass sie in proximalen Tubuli speziell vermehrt DUSP26 produzierten, einen beeindruckenden Schutz vor sowohl medikamenteninduzierter als auch ischämischer Schädigung, mit besserer Nierenfunktion und weniger Gewebeschaden. Diese Gewinn‑ und Verlust‑Funktions‑Experimente machen DUSP26 gemeinsam zu einem starken Wächter der Tubulusintegrität.
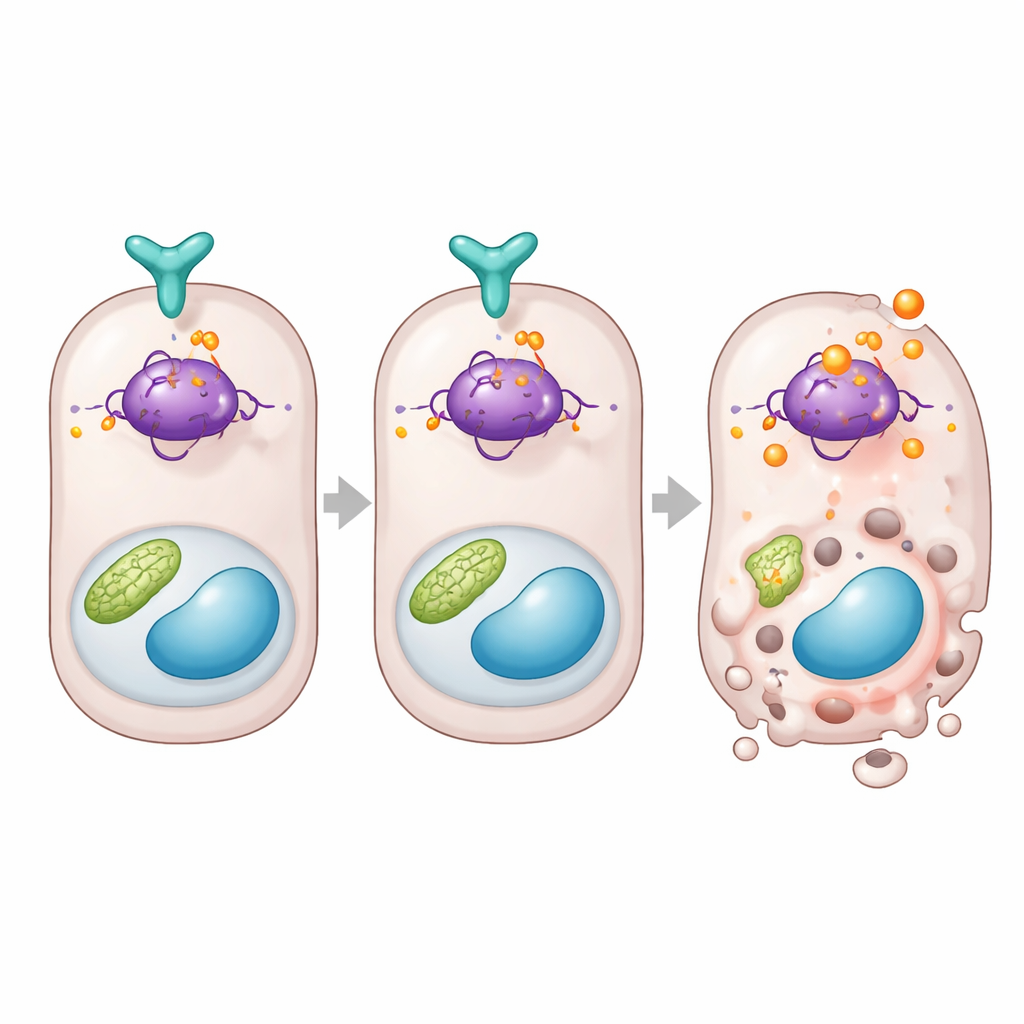
Eine direkte Bremse für das zelltodentscheidende Protein
Tiefer gehend verbindet die Studie DUSP26 mit p53, einem bekannten Protein, das entscheidet, ob eine gestresste Zelle sich repariert oder sich selbst zerstört. Das Team fand heraus, dass bei niedrigem DUSP26 in menschlichen Daten oder in Mausnierenn p53‑gesteuerte Todesgene wie Bax und Puma hochreguliert waren. In Zellen band DUSP26 physikalisch an p53 und entfernte selektiv eine Phosphatgruppe an einer spezifischen Stelle des Proteins, bekannt als Serin 312 bei Mäusen. War DUSP26 nicht vorhanden, wurde diese Stelle stark modifiziert, die p53-Aktivität stieg und todfördernde Gene wurden eingeschaltet. Um die Bedeutung dieser Stelle zu belegen, erzeugten die Autoren Nierenzellen und Mäuse mit einer Version von p53, die an dieser Position nicht mehr modifiziert werden konnte. Diese Zellen und Tiere waren deutlich widerstandsfähiger gegen Nierenschädigung, mit weniger sterbenden Tubuluszellen und geringerer Organfehlfunktion, obwohl andere Teile des p53-Systems intakt blieben.
Von molekularer Erkenntnis zu möglichen Therapien
Insgesamt skizziert die Arbeit eine klare Ereigniskette: Nierenstress erhöht die DNA-Methylierung des DUSP26-Gens und reduziert das DUSP26-Protein in proximalen Tubuli. Mit weniger verfügbarem DUSP26 bleibt p53 an einer kritischen Stelle phosphoryliert, wird aktiver und treibt die Expression von Genen voran, die Zellen in Richtung Tod lenken. Die Wiederherstellung von DUSP26 oder das Nachahmen seiner Wirkung an genau dieser Stelle dämpft dieses Todesprogramm und erhält Nierengewebe. Für Patientinnen und Patienten bedeutet das, dass sorgfältig entwickelte Medikamente oder gentherapieartige Ansätze, die die DUSP26-Aktivität erhöhen oder speziell die Phosphorylierung von p53 an dieser Stelle reduzieren, eines Tages helfen könnten, Nieren bei Operationen, Chemotherapie oder schweren Erkrankungen zu schützen und möglicherweise auch andere Organe vor ähnlichen Formen akuter Schädigung zu bewahren.
Zitation: Fu, Y., Xiang, Y., Han, Y. et al. DUSP26 protects against acute kidney injury by dephosphorylating p53 at serine 312. Nat Commun 17, 3208 (2026). https://doi.org/10.1038/s41467-026-69688-3
Schlüsselwörter: akutes Nierenversagen, Nierentubuluszellen, p53-Signalgebung, epigenetische Regulation, Zellsterben