Clear Sky Science · en
DUSP26 protects against acute kidney injury by dephosphorylating p53 at serine 312
Why kidney stress matters to everyone
Acute kidney injury is a sudden drop in kidney function that can strike during major surgery, severe infection, dehydration, or cancer treatment. It is common in hospitals and raises the risk of needing dialysis, staying in the ICU longer, and later developing chronic kidney disease. Yet doctors still have no targeted medicine that directly protects the kidney’s working cells. This study uncovers a built‑in protective switch inside those cells and shows how flipping that switch on or off can dramatically change whether kidneys are damaged or spared.
A hidden defender inside kidney cells
The authors focused on the tiny tubes inside the kidney that filter and fine‑tune our blood. By mining several large human datasets with modern machine‑learning tools, they pinpointed a gene called DUSP26 that consistently showed up at lower levels in people with acute kidney injury than in those with healthy kidneys. When they examined human biopsy samples, they found that proximal tubule cells—the cells that do most of the kidney’s heavy lifting—had sharply reduced amounts of the DUSP26 protein during injury, and this loss tracked closely with worse tissue damage and poorer kidney function. This suggested that DUSP26 normally helps these cells withstand stress.
How the kidney’s shield is switched off
To understand why DUSP26 levels fall, the team turned to mouse models of two common forms of kidney stress: a chemotherapy drug that is toxic to the kidney and a period of blocked blood flow followed by reperfusion. In both situations, DUSP26 levels in the kidney dropped in parallel with rising cell death. They traced this loss to chemical tags, called DNA methylation, laid down across the DUSP26 gene’s control region. These tags act like pieces of tape over a light switch, preventing the gene from being turned on. Experiments in cultured kidney cells showed that injury increased methylation and shut down DUSP26, and that enzymes responsible for adding these tags were drawn to the gene’s promoter. In mice with ischemia‑reperfusion injury, deep sequencing confirmed that many sites within the same promoter region became heavily methylated.
Turning protection off and on in animals
The researchers then asked what happens if this molecular shield is removed or reinforced. In cell culture, blocking DUSP26 with genetic tools or a small‑molecule drug led to more activation of caspase‑3, a key executioner of programmed cell death, after toxic or low‑oxygen stress. Boosting DUSP26 had the opposite effect, cutting cell death. In mice, pretreating animals with the DUSP26 inhibitor before kidney injury caused higher blood waste levels, more microscopic damage, and more dying tubule cells. In contrast, mice engineered to overproduce DUSP26 specifically in proximal tubules showed striking protection from both drug‑induced and ischemic injury, with better kidney function and less tissue damage. These gain‑ and loss‑of‑function experiments together reveal DUSP26 as a powerful guardian of tubular integrity.
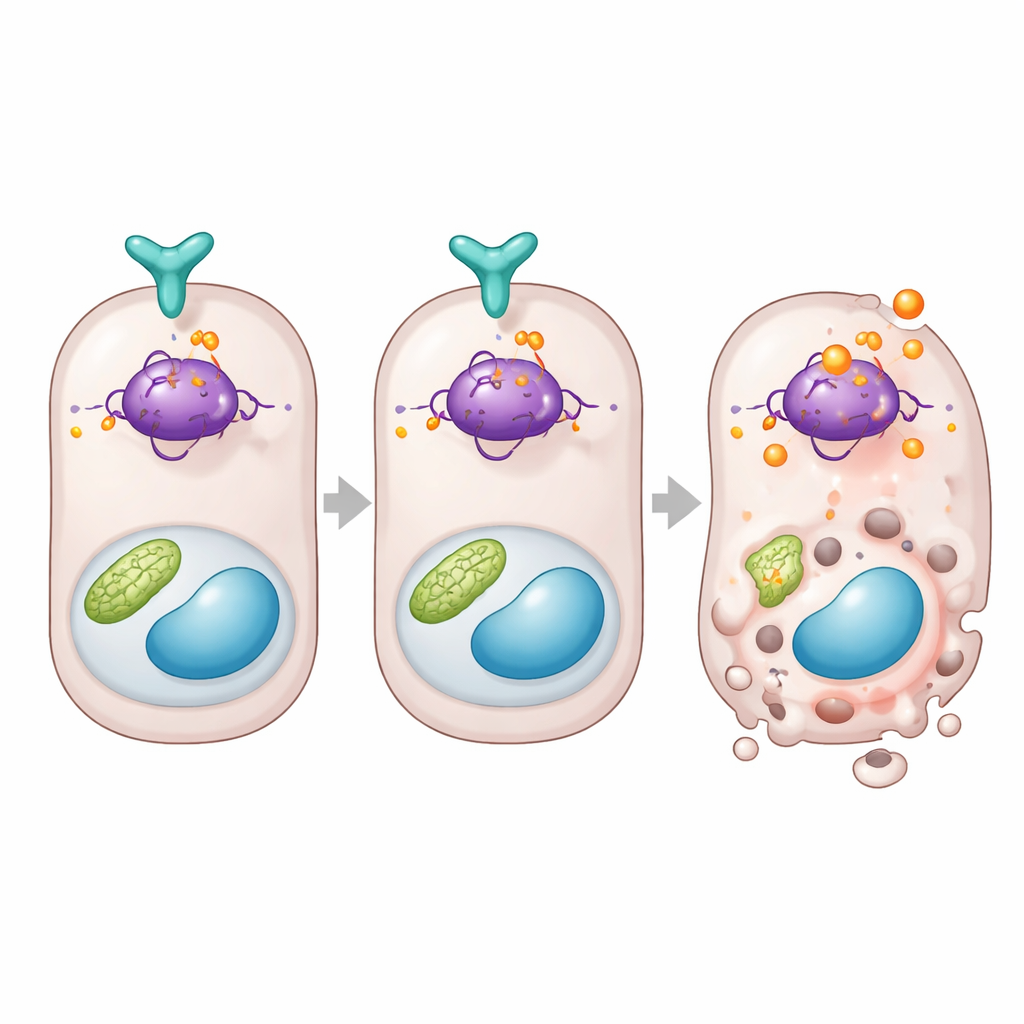
A direct brake on the cell’s death‑deciding protein
Digging deeper, the study connects DUSP26 to p53, a famous protein that decides whether a stressed cell repairs itself or self‑destructs. The team found that when DUSP26 was low in human data or mouse kidneys, p53‑controlled death genes such as Bax and Puma were turned up. In cells, DUSP26 physically bound to p53 and selectively removed a phosphate group from one specific site on the protein, known as serine 312 in mice. When DUSP26 was absent, this site became heavily modified, p53 activity rose, and death‑promoting genes switched on. To prove this site’s importance, the authors rebuilt kidney cells and mice with a version of p53 that could no longer be modified at that spot. Those cells and animals were far more resistant to kidney injury, with fewer dying tubule cells and less organ dysfunction, even though other parts of the p53 system remained intact.
From molecular insight to future therapies
Taken together, the work outlines a clear chain of events: kidney stress increases DNA methylation on the DUSP26 gene, lowering DUSP26 protein in proximal tubules. With less DUSP26 available, p53 remains phosphorylated at a critical site, becomes more active, and drives the expression of genes that push cells toward death. Restoring DUSP26, or mimicking its action on that single site, dampens this death program and preserves kidney tissue. For patients, this suggests that carefully designed drugs or gene‑targeted approaches that enhance DUSP26 activity—or specifically reduce phosphorylation of p53 at this site—could one day help shield kidneys during surgery, chemotherapy, or severe illness, and perhaps protect other organs from similar forms of acute injury.
Citation: Fu, Y., Xiang, Y., Han, Y. et al. DUSP26 protects against acute kidney injury by dephosphorylating p53 at serine 312. Nat Commun 17, 3208 (2026). https://doi.org/10.1038/s41467-026-69688-3
Keywords: acute kidney injury, kidney tubule cells, p53 signaling, epigenetic regulation, cell death