Clear Sky Science · it
Direttamente dal seme: una struttura proteica a risoluzione atomica ottenuta ab initio con MicroED
Osservare i più piccoli mattoni della vita
Le proteine sono le piccole macchine che mantengono funzionanti le cellule viventi, ma per comprendere davvero il loro funzionamento gli scienziati devono vedere gli atomi con chiarezza straordinaria. Questo articolo descrive come i ricercatori abbiano utilizzato un potente metodo basato sugli elettroni per determinare una struttura proteica a livello atomico partendo direttamente da semi polverizzati, senza fare affidamento su modelli strutturali preesistenti. Il loro successo offre un nuovo riferimento per lo studio di proteine piccole o difficili che resistono alle tecniche tradizionali.
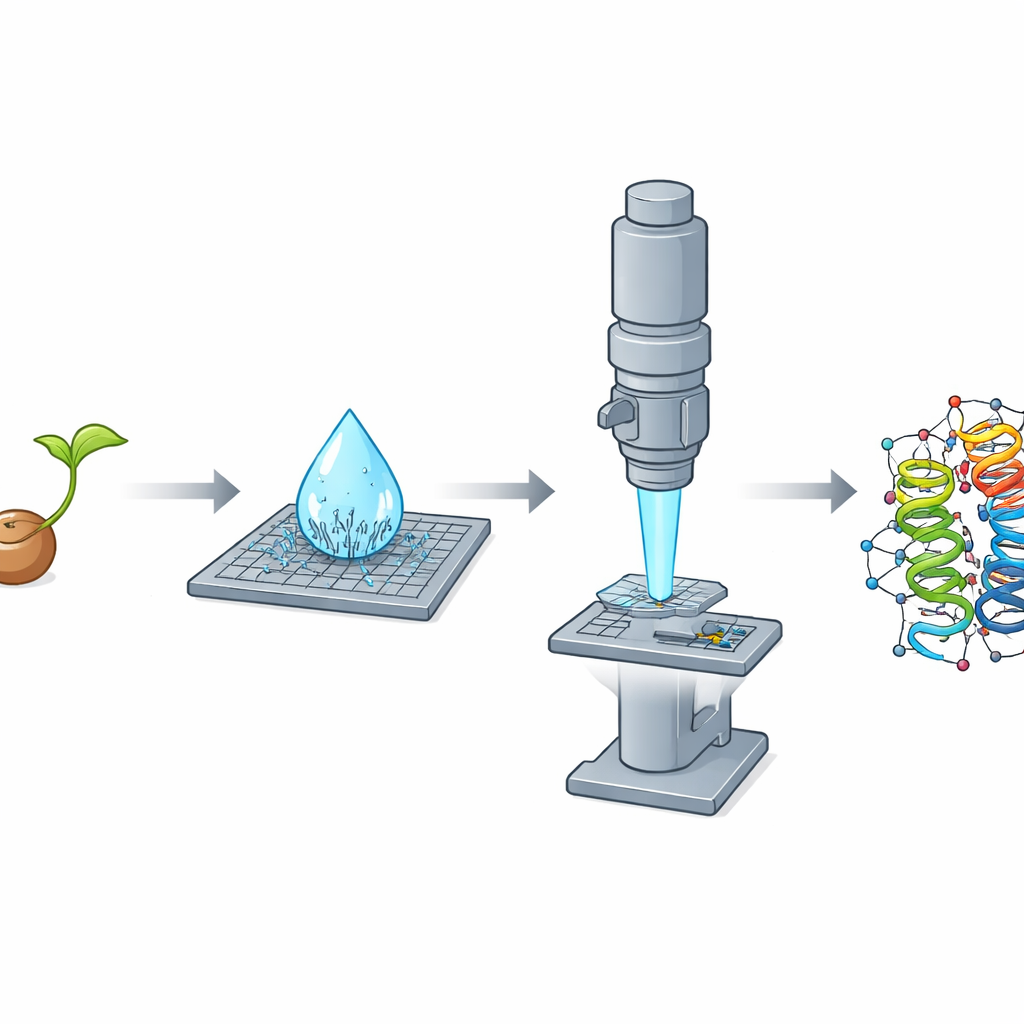
Perché i cristalli minuscoli sono un grande problema
Per decenni, la cristallografia a raggi X è stata lo strumento principale per mappare le strutture proteiche a risoluzione atomica. Tuttavia richiede cristalli relativamente grandi e altamente ordinati—spesso dell’ordine di decine o centinaia di micrometri. Molte proteine importanti formano naturalmente solo cristalli minuscoli, molto più piccoli di questi, e quindi sfuggono alle maglie dei metodi standard. Un altro approccio diffuso, la criomicroscopia elettronica single-particle, fatica con proteine più piccole, specialmente quelle vicine alle dimensioni tipiche delle proteine umane. Considerate insieme, queste limitazioni lasciano una vasta porzione della biologia senza una mappatura strutturale.
Usare elettroni invece dei raggi X
La diffrazione elettronica da microcristalli, o MicroED, affronta questo problema usando elettroni invece dei raggi X. Gli elettroni interagiscono molto più fortemente con la materia, permettendo agli scienziati di estrarre informazioni strutturali dettagliate da cristalli migliaia di volte più piccoli in volume rispetto a quelli richiesti per studi con raggi X. In questo studio il gruppo si è concentrato sul crambin, una proteina piccola presente nei semi della pianta oleaginosa Crambe abyssinica. Il crambin è da tempo un banco di prova per metodi cristallografici avanzati, con lavori precedenti che usando raggi X e neutroni hanno spinto la risoluzione a livelli straordinari. Tuttavia, un’indagine completa e ab initio del crambin con MicroED non era ancora stata realizzata del tutto.
Una scoperta fortunata in una goccia che si asciuga
Mentre purificavano il crambin da semi triturati con una semplice procedura a base di etanolo, i ricercatori hanno osservato qualcosa di sorprendente: quando una goccia da microlitro della soluzione si asciugava su un vetrino, in pochi secondi produceva spontaneamente una fitta pioggia di nanocristalli aghiformi. Questi filamenti si sono rivelati poco adatti per i raggi X ma quasi ideali per la diffrazione elettronica, perché erano estremamente sottili lungo una dimensione e quindi trasparenti al fascio di elettroni. Il gruppo ha inoltre coltivato cristalli tradizionali, a forma di blocco, che eccellevano negli esperimenti con raggi X ma erano troppo spessi per MicroED, anche dopo la frantumazione. Questo netto capovolgimento—aghi cattivi per i raggi X ma ottimi per gli elettroni, blocchi il contrario—ha messo in evidenza come la forma e lo spessore del cristallo possano determinare quale metodo sia vincente.
Combinare molti aghi per ottenere un quadro chiaro
C’era però un problema: i cristalli aghiformi tendevano a disporsi nella stessa orientazione sulla griglia del microscopio, per cui qualsiasi singolo cristallo forniva una vista incompleta e sbilanciata della proteina. Per superare questo limite, i ricercatori hanno adottato una strategia seriale, raccogliendo piccole porzioni di dati da 58 cristalli diversi e unendole in un unico dataset ampio. Poiché la qualità dei dati dipendeva fortemente dalla direzione di osservazione, hanno utilizzato una fase di elaborazione specializzata che riconosce e elimina misure deboli e rumorose nelle direzioni scarsamente campionate, mantenendo invece i segnali forti dove i cristalli diffrangono meglio. Questo trattamento ha prodotto un dataset di altissima qualità che ha raggiunto una risoluzione migliore dell’ångström, sufficientemente ricco da rivelare singoli atomi, inclusi molti idrogeni.
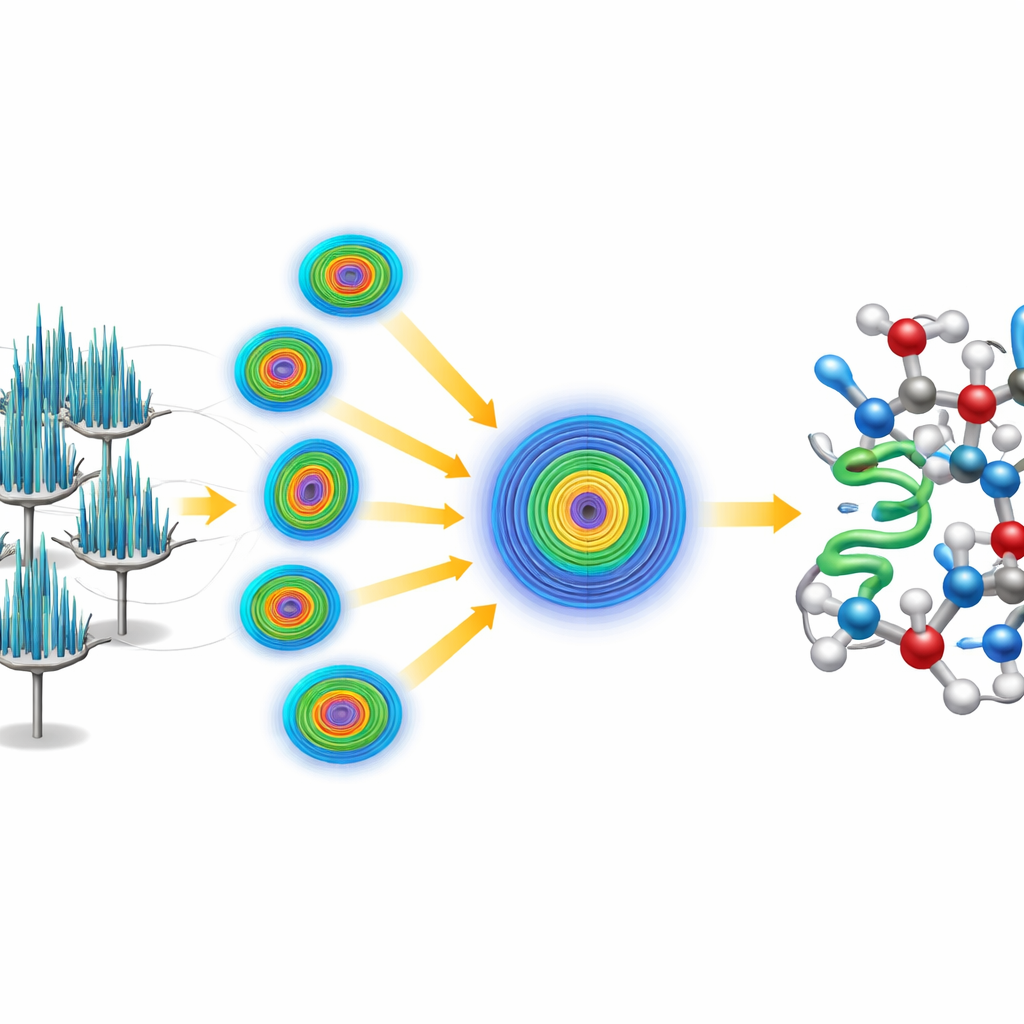
Risoluzione della struttura da zero
Invece di appoggiarsi a un modello esistente di crambin, il team si è sfidato a risolvere la struttura “ab initio”, ossia dai primi principi. Hanno iniziato con un piccolo elica generica di cinque amminoacidi—essenzialmente un breve frammento proteico idealizzato non correlato alla struttura nota del crambin. Usando software consolidato, hanno individuato dove questo frammento potesse plausibilmente collocarsi nel motivo ripetuto del cristallo, quindi hanno applicato una procedura iterativa di affinamento che ha progressivamente migliorato la mappa derivata dagli elettroni. La mappa raffinata è diventata sufficientemente chiara perché software automatici potessero tracciare l’intera catena di crambin senza intervento umano, producendo un modello completo ad alta risoluzione. I dati erano così dettagliati che i ricercatori hanno potuto distinguere e quantificare due varianti naturali del crambin che differiscono in appena due posizioni della catena.
Cosa significa per gli studi futuri
Il lavoro dimostra che, con una raccolta dati accurata e un’elaborazione che tenga conto dell’anisotropia, è possibile raggiungere una vera risoluzione atomica e risolvere una struttura proteica da zero usando solo nanocristalli formatisi spontaneamente e attrezzature microscopiche elettroniche standard. Il crambin ora costituisce un dataset di riferimento pubblico che altri scienziati possono usare per testare nuovi metodi di analisi, perfezionare i modelli di scattering elettronico ed esplorare fenomeni come i legami a idrogeno e la distribuzione di carica. Per i non specialisti, il punto chiave è che potenti nuovi strumenti basati sugli elettroni stanno estendendo il campo della biologia strutturale, rendendo sempre più fattibile visualizzare i dettagli più minuti di proteine un tempo ritenute troppo piccole, fragili o non convenzionali per essere studiate a livello atomico.
Citazione: Vasireddy, P.C.R., Low-Beer, T., Spoth, K.A. et al. Direct from the seed: an atomic resolution protein structure by ab initio MicroED. Nat Commun 17, 2759 (2026). https://doi.org/10.1038/s41467-026-69601-y
Parole chiave: MicroED, struttura proteica, crambin, diffrazione elettronica, nanocristalli